СПЕКТРАЛЬНЫЙ АНАЛИЗ (при помощи спектров испускания) имеет применение почти во всех отраслях хозяйства. Широко применяется в металлопромышленности для быстрого анализа железа, стали, чугуна, а также различных специальных сталей и готовых металлических изделий, для установления чистоты легких, цветных и драгоценных металлов. Большое применение имеет спектральный анализ в геохимии при изучении состава полезных ископаемых. В химической промышленности и близких к ней отраслях спектральный анализ служит для установления чистоты выпускаемой и применяемой продукции, для анализа катализаторов, различных остатков, осадков, мутей и промывных вод; в медицине - для открытия металлов в различных органических тканях. Ряд специальных задач, трудно разрешаемых или вовсе не разрешимых иным путем, решается при помощи спектрального анализа быстро и точно. Сюда относится, например, распределение металлов в сплавах, исследование в сплавах и минералах сульфидных и других включений; такого рода исследования иногда обозначаются термином локальный анализ .
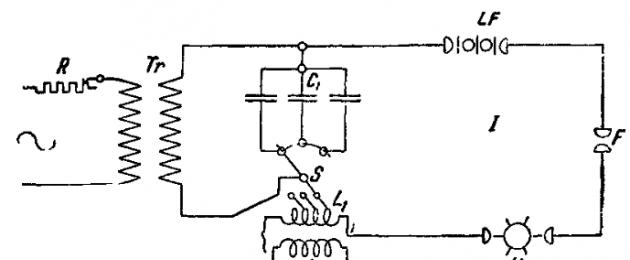
Выбор того или другого типа спектрального аппарата с точки зрения достаточности его дисперсии производится в зависимости от цели и задач спектрального анализа. Для исследования платиновых металлов (Ru, Rh, Pd, Os, Ir, Pt), а также Fe, Co, Ni, Сг, V, Mo, W, Ti, Mn, Zr, Re, Nb и Та наиболее пригодны кварцевые спектрографы с большей дисперсией, дающие для длин волн 4000-2200 Ӑ полоску спектра длиной по крайней мере 22 см. Для остальных элементов м. б. применены аппараты, дающие спектры длиной 7-15 см. Спектрографы со стеклянной оптикой в общем имеют меньшее значение. Из них удобны комбинированные приборы (например, фирмы Гильгера и Фюсса), которые по желанию можно применять в качестве спектроскопа и спектрографа. Для получения спектров применяются следующие источники энергии. 1) Пламя горящей смеси - водорода и кислорода, смеси кислорода и светильного газа, смеси кислорода и ацетилена или наконец воздуха и ацетилена. В последнем случае температура источника света доходит до 2500-3000°С. Пламя наиболее всего пригодно для получения спектров щелочных и щелочноземельных металлов, а также для таких элементов, как Сu, Hg и Тl. 2) Вольтова дуга . а) Обычная, гл. обр. постоянного тока, силой 5-20 А. С большим успехом она применяется для качественного анализа трудно сплавляемых минералов, которые вводятся в дугу в виде кусочков или тонко растертых порошков. Для количественного анализа металлов применение обычной вольтовой дуги имеет очень существенный недостаток, заключающийся в том, что поверхность анализируемых металлов покрывается пленкой окиси и горение дуги становится в конце концов неравномерным. Температура вольтовой дуги доходит до 5000-6000°С. б) Прерывистая дуга (Abreissbogen) постоянного тока силой 2-5 А при напряжении около 80 V. При помощи специального приспособления горение дуги прерывается 4-10 раз в сек. Этот способ возбуждения уменьшает окисление поверхности анализируемых металлов. При более высоком напряжении - до 220 V и силе тока 1-2 А - прерывистая дуга может применяться также и для анализа растворов. 3) Искровые разряды , получаемые при помощи индукционной катушки или, чаще, трансформатора постоянного или (предпочтительнее) переменного тока мощностью до 1 kW, дающего во вторичной цепи 10000-30000 V. Применяются три типа разрядов, а) Искровые разряды без емкости и индуктивности во вторичной цепи, называемые иногда дугой высокого напряжения (Hochspannungsbogen). Анализ жидкостей и расплавленных солей при помощи таких разрядов отличается большой чувствительностью. б) Искровые разряды с емкостью и индуктивностью во вторичной цепи, часто называемые также конденсированными искрами , представляют собой более универсальный источник энергии, пригодный для возбуждения спектров почти всех элементов (кроме щелочных металлов), а также газов. Схема включения дана на фиг. 1,

где R - реостат в первичной цепи, Тr- трансформатор переменного тока, С 1 - емкость во вторичной цепи I, S - переключатель для изменения индуктивности L 1 , U - синхронный прерыватель, LF - искрогаситель, F - рабочий искровой промежуток. В резонанс ко вторичной цепи I при помощи индуктивности и переменной емкости С 2 настраивается вторичная цепь II; признаком наличия резонанса является наибольшая сила тока, показываемая миллиамперметром А. Назначение вторичной цепи II синхронного прерывателя U и искрогасителя LF - делать электрические разряды возможно однообразными как по характеру, так и по числу в течение определенного промежутка времени; при обычных работах такие добавочные приспособления не вводятся.
При исследованиях металлов во вторичной цепи применяется ёмкость 6000-15000 см и индуктивность до 0,05-0,01 Н. Для анализа жидкостей во вторичную цепь иногда вводится водяной реостат с сопротивлением до 40000 Ом. Газы исследуются без индуктивности с небольшой емкостью. в) Разряды токов Тесла, которые осуществляются при помощи схемы, изображенной на фиг. 2,

где V - вольтметр, А - амперметр, Т - трансформатор, С - емкость, Т-Т - трансформатор Тесла, F - искровой промежуток, куда вводится анализируемое вещество. Токи Тесла применяются для исследований веществ, которые имеют невысокую точку плавления: различных растительных и органических препаратов, осадков на фильтрах и т. п. При спектральном анализе металлов в случае большого их количества они обычно сами являются электродами, причем им придается какая-либо форма, например, из указанных на фиг. 3,

где а - электрод из анализируемой толстой проволоки, b - из жести, с - согнутая тонкая проволока, d - диск, отрезанный от толстого цилиндрического стержня, е - форма, выпиливаемая из больших кусков литья. При количественном анализе необходимо иметь всегда одинаковую форму и размеры подвергающейся действию искр поверхности электродов. При небольшом количестве анализируемого металла можно воспользоваться оправой из какого-либо чистого металла, например, из золота и платины, в которой укрепляется анализируемый металл, как показано на фиг. 4.
Для введения в источник света растворов предложено довольно много способов. При работе с пламенем применяется распылитель Люндегорда, схематически изображенный на фиг. 5 вместе со специальной горелкой.

Продуваемый через распылитель ВС воздух захватывает испытуемую жидкость, наливаемую в количестве 3 -10 см 3 в углубление С, и в виде тонкой пыли относит ее в горелку А, где происходит смешение с газом. Для введения растворов в дугу, а также в искру применяются чистые угольные или графитовые электроды, на одном из которых делается углубление. Необходимо, однако, отметить, что очень трудно приготовить угли совершенно чистыми. Применяемые для очистки способы - попеременное кипячение в соляной и плавиковой кислотах, а также прокаливание в атмосфере водорода до 2500-3000°С - не дают углей, свободных от примесей, остаются (хотя и следы) Са, Mg, V, Ti, Al, Fe, Si, В. Удовлетворительной чистоты получаются также угли путем прокаливания их на воздухе при помощи электрического тока: через угольный стержень диаметром 5 мм пропускается ток силой около 400 А, и достигаемое таким путем сильное накаливание (до 3 000°С) оказывается достаточным для того, чтобы в течение нескольких секунд большинство загрязняющих угли примесей улетучилось. Существуют также такие способы введения растворов в искру, где сам раствор является нижним электродом, и искра проскакивает на его поверхность; другим электродом может служить какой-либо чистый металл. Примером такого устройства может служить изображенный на фиг. 6 жидкостный электрод Герляха.

Углубление, куда наливается испытуемый раствор, облицовывается платиновой фольгой или покрывается толстым слоем позолоты. На фиг. 7 изображен аппарат Хитчена, служащий также для введения растворов в искру.

Из сосуда А испытуемый раствор слабой струей поступает через трубку В и кварцевую насадку С в сферу действия искровых разрядов. Нижний электрод, впаянный в стеклянную трубку, прикрепляется к аппарату при помощи каучуковой трубки Е. Насадка С, изображенная на фиг. 7 отдельно, имеет с одной стороны вырез для стенания раствора. D - стеклянный предохранительный сосуд, в котором делается круглое отверстие для выхода ультрафиолетовых лучей. Сосуд этот удобнее делать кварцевым без отверстия. К верхнему электроду F, графитовому, угольному или металлическому, также приспосабливается предохраняющая от брызг пластинка. Для «дуги высокого напряжения», сильно накаливающей анализируемые вещества, Герлях при работе с растворами применяет электроды с охлаждением, как это схематически показано на фиг. 8.

На толстой проволоке (диаметром 6 мм) укрепляется при помощи пробки К стеклянная воронка G, куда помещаются кусочки льда. На верхнем конце проволоки укрепляется круглый железный электрод Е диаметром 4 см и высотой 4 см, на который накладывается платиновая чашечка Р; последняя должна легко сниматься для очистки. Верхний электрод также д. б. толстым во избежание расплавления. При анализе небольших количеств веществ - осадков на фильтрах, различных порошков и т. д. - можно пользоваться приспособлением, изображенным на фиг. 9.
Из испытуемого вещества и фильтровальной бумаги делается комочек, смачивается для лучшей проводимости раствором, например, NaCl, помещается на нижний электрод, состоящий иногда из чистого кадмия, заключенного в кварцевой (хуже стеклянной) трубочке; верхний электрод также является каким-либо чистым металлом. Для таких же анализов при работе с токами Тесла применяется специальная конструкция искрового промежутка, изображенная на фиг. 10 а и б.

В круглом шарнире К укрепляется в нужном положении алюминиевая пластинка Е, на которую накладывается стеклянная пластинка G, а на последнюю - препарат Р на фильтровальной бумаге F. Препарат смачивается какой-либо кислотой или раствором соли. Вся эта система представляет небольшой конденсатор. Для исследования газов применяются закрытые стеклянные или кварцевые сосуды (фиг. 11).

Для количественного анализа газов удобно пользоваться золотыми или платиновыми электродами, линии которых можно применить для сравнения. Почти все из упомянутых выше приспособлений для введения веществ в искру и дугу при работе укрепляются в специальных штативах. Примером может являться штатив Грамона, изображенный на фиг. 12:

при помощи винта D электроды одновременно раздвигаются и сдвигаются; винт Е служит для передвигания верхнего электрода параллельно оптической скамье, а винт С - для боковых поворотов нижнего электрода; для боковых поворотов всей верхней части штатива служит винт В; наконец при помощи винта А можно поднимать или опускать всю верхнюю часть штатива; Н - подставка для горелок, стаканов и пр. Выбор источника энергии для той или иной цели исследования можно сделать, руководствуясь следующей примерной таблицей.

Качественный анализ . При качественном спектральном анализе открытие какого-либо элемента зависит от многих факторов: от характера определяемого элемента, источника энергии, разрешающей способности спектрального аппарата, а также от чувствительности фотографических пластинок. Относительно чувствительности анализа можно сделать следующие указания. При работе с искровыми разрядами в растворах можно открывать 10 -9 -10 -3 %, а в металлах 10 -2 -10 -4 % исследуемого элемента; при работе с вольтовой дугой пределы открытия лежат около 10 -3 %. Абсолютное количество, которое м. б. открыто при работе с пламенем, составляет 10 -4 -10 -7 г, а при искровых разрядах 10 -6 -10 -8 г исследуемого элемента. Наибольшая чувствительность открытия относится к металлам и металлоидам - В, Р, С; меньше чувствительность для металлоидов As, Se и Те; галоиды, а также S, О, N в их соединениях совсем не м. б. открыты и м. б. открыты лишь в некоторых случаях в газовых смесях.
Для качественного анализа наибольшее значение имеют «последние линии», и при анализе задача заключается в наиболее точном определении длин волн спектральных линий. При визуальных исследованиях длины волн отсчитываются по барабану спектрометра; эти измерения можно считать лишь приблизительными, так как точность составляет обычно ±(2-З) Ӑ и в таблицах Кайзера этому интервалу ошибок могут отвечать около 10 спектральных линий, принадлежащих различным элементам, для λ 6000 и 5000 Ӑ и около 20 спектральных линий для λ ≈ 4000 Ӑ. Гораздо точнее определяется длина волн при спектрографическом анализе. В этом случае на спектрограммах при помощи измерительного микроскопа измеряется расстояние между линиями с известной длиной волны и определяемой; по формуле Гартмана находится длина волны последней. Точность таких измерений при работе с прибором, дающим полоску спектра длиной около 20 см, составляет ± 0,5 Ӑ для λ ≈ 4000 Ӑ, ± 0,2 Ӑ для λ ≈ 3000 Ӑ и ± 0,1 Ӑ для λ ≈ 2500 Ӑ. По длине волны в таблицах находят соответствующий элемент. Расстояние между линиями при обычных работах измеряется с точностью до 0,05-0,01 мм. Этот прием иногда удобно комбинировать со съемками спектров с так называемыми заслонками Гартмана, два типа которых изображены на фиг. 13, а и b; при помощи их щель спектрографа можно делать различной высоты. Фиг. 13, с схематически изображает случай качественного анализа вещества X - установление в нем элементов А и В. Спектры фиг. 13, d показывают, что в веществе Y кроме элемента А, линии которого обозначены буквой G, имеется примесь, линии которой обозначены z. При помощи этого приема в простых случаях можно выполнить качественный анализ, не прибегая к промеру расстояний между линиями.

Количественный анализ . Для количественного спектрального анализа наибольшее значение имеют линии, обладающие возможно большей концентрационной чувствительностью dI/dK, где I - интенсивность линии, а К - концентрация дающего ее элемента. Чем больше концентрационная чувствительность, тем точнее анализ. С течением времени разработан целый ряд методов количественного спектрального анализа. Эти методы следующие.
I. Спектроскопические методы (без фотографической съемки) почти все являются фотометрическими методами. Сюда относятся: 1) Метод Барратта . Одновременно возбуждаются спектры двух веществ - испытуемого и стандартного - видные в поле зрения спектроскопа рядом, один над другим. Ход лучей изображен на фиг. 14,

где F 1 и F 2 - два искровых промежутка, свет от которых проходит через призмы Николя N 1 и N 2 , поляризующие лучи во взаимно перпендикулярных плоскостях. При помощи призмы D лучи попадают в щель S спектроскопа. В его зрительной трубе помещается третья призма Николя - анализатор, - вращая которую добиваются одинаковой интенсивности двух сравниваемых линий. Предварительно при исследованиях стандартов, т. е. веществ с известным содержанием элементов, устанавливается зависимость между углом поворота анализатора и концентрацией, и по этим данным вычерчивается диаграмма. При анализе по углу поворота анализатора из этой диаграммы находится искомое процентное содержание. Точность метода ±10 %. 2) . Принцип метода заключается в том, что лучи света после призмы спектроскопа проходят через призму Волластона, где расходятся на два пучка и поляризуются во взаимно перпендикулярных плоскостях. Схема хода лучей показана на фиг. 15,

где S - щель, Р - призма спектроскопа, W - призма Волластона. В поле зрения получаются два спектра B 1 и В 2 , лежащие рядом, друг над другом; L - лупа, N - анализатор. Если вращать призму Волластона, то спектры будут передвигаться относительно друг друга, что позволяет совместить какие-либо две их линии. Например, если анализируется железо, содержащее ванадий, то совмещается линия ванадия с какой-либо близлежащей одноцветной линией железа ; затем, поворачивая анализатор, добиваются одинаковой яркости этих линий. Угол поворота анализатора, как и в предыдущем методе, является мерой концентрации искомого элемента. Метод особенно пригоден для анализа железа, спектр которого имеет много линий, что позволяет всегда найти линии, пригодные для исследований. Точность метода ± (3-7)%. 3) Метод Оккиалини . Если расположить электроды (например, анализируемые металлы) горизонтально и проектировать изображен из источника света на вертикальную щель спектроскопа, то как при искровых, так и при дуговых разрядах линии примесей м. б. открыты в зависимости от концентрации на большем или меньшем расстоянии от электродов. Источник света проектируется на щель при помощи специальной линзы, снабженной микрометрическим винтом. При анализе эта линза передвигается и вместе с ней передвигается изображение источника света до тех пор, пока какая-либо линия примеси в спектре исчезнет. Мерой концентрации примеси является отсчет по шкале линзы. В настоящее время этот метод разработан также и для работ с ультрафиолетовой частью спектра. Надо отметить, что таким же способом освещения щели спектрального аппарата пользовался Локиер и им был разработан метод количественного спектрального анализа, т. н. метод «длинных и коротких линий». 4) Прямое фотометрирование спектров . Описанные выше методы носят название визуальных. Люндегорд вместо визуальных исследований пользовался для измерения интенсивности спектральных линий фотоэлементом. Точность определения щелочных металлов при работе с пламенем достигала ± 5%. При искровых разрядах этот способ неприменим, так как они менее постоянны, чем пламя. Существуют также способы, основанные на изменении индуктивности во вторичной цепи, а также использующие искусственное ослабление света, попадающего в спектроскоп, до исчезновения в поле зрения исследуемых спектральных линий.
II. Спектрографические методы . При этих методах исследуются фотографические снимки спектров, причем мерой интенсивности спектральных линий является почернение, даваемое ими на фотографической пластинке. Интенсивность оценивается или глазом, или фотометрически.
А . Методы без применения фотометрии . 1) Метод последних линий . При изменении концентрации какого-либо элемента в спектре изменяется число его линий, что дает возможность при неизменных условиях работы судить о концентрации определяемого элемента. Фотографируется ряд спектров веществ с известным содержанием интересующего компонента, на спектрограммах определяется число его линий и составляются таблицы, в которых указывается, какие линии видны при данных концентрациях. Эти таблицы служат дальше для аналитических определений. При анализе на спектрограмме определяется число линий интересующего элемента и по таблицам находится процентное содержание, причем метод дает не однозначную его цифру, а границы концентраций, т. е. «от-до». Наиболее достоверно возможно различить концентрации, отличающиеся друг от друга в 10 раз, например, от 0,001 до 0,01%, от 0,01 до 0,1% и т. д. Аналитические таблицы имеют значение лишь для вполне определенных условий работы, которые в различных лабораториях могут очень сильно различаться; кроме того, требуется тщательное соблюдение постоянства условий работы. 2) Метод сравнительных спектров . фотографируется несколько спектров анализируемого вещества А + х% В, в котором определяется содержание х элемента В, и в промежутках между ними на той же фотографической пластинке -спектры стандартных веществ А + а% В, А + b% В, А + с% В, где а, b, с - известное процентное содержание В. На спектрограммах по интенсивности линий В определяется, между какими концентрациями заключается значение х. Критерием постоянства условий работы является равенство интенсивности на всех спектрограммах какой-либо близлежащей линии А. При анализе растворов в них добавляется одинаковое количество какого-либо элемента, дающего линию близко к линиям В, и тогда о постоянстве условий работы судят по равенству интенсивности этих линий. Чем меньше разница между концентрациями а, Ь, с, … и чем точнее достигнуто равенство интенсивности линий А, тем точнее анализ. А. Райс, например, применял концентрации а, b, с, ... , относящиеся друг к другу, как 1: 1,5. К методу сравнительных спектров примыкает метод «подбора концентраций» (Testverfahren) по Гюттигу и Турнвальду, применимый только к анализу растворов. Он заключается в том, что если в двух растворах, содержащих а% А и х% А (х больше или меньше а), что сейчас же можно определить по их спектрам, то прибавляют в какой-либо из этих растворов такое количество n элемента А, чтобы интенсивность его линий на обоих спектрах стала одинаковой. Тем самым определится концентрация х, которая будет равна (а ± n)%. Можно также прибавить в анализируемый раствор какой-либо другой элемент В до равенства интенсивности определенных линий А и В и по количеству В оценить содержание А. 3) Метод гомологических пар . В спектре вещества А + а% В линии элементов А и В не являются одинаково интенсивными и, если этих линий достаточное количество, можно найти две такие линии А и В, интенсивность которых будет одинакова. Для другого состава А + b% В одинаковыми по интенсивности будут другие линии А и В и т. д. Эти две одинаковые линии называются гомологическими парами. Концентрации В, при которых осуществляется та или иная гомологическая пара, называются фиксирующими пунктами этой пары. Для работы по этому методу требуется предварительное составление таблиц гомологических пар при помощи веществ известного состава. Чем полнее таблицы, т. е. чем больше они содержат гомологических пар с фиксирующими пунктами, отличающимися как можно меньше друг от друга, тем точнее анализ. Этих таблиц составлено довольно большое количество, причем они могут иметь применение в любой лаборатории, т. к. точно известны условия разрядов при их составлении и эти условия м. б. совершенно точно воспроизведены. Достигается это при помощи следующего простого приема. В спектре вещества А + а% В выбираются две линии элемента А, интенсивность которых очень сильно меняется в зависимости от величины самоиндукции во вторичной цепи, именно одна дуговая (принадлежащая нейтральному атому) и одна искровая линия (принадлежащая иону). Эти две линии называются фиксирующей парой . Путем подбора величины самоиндукции линии этой пары делаются одинаковыми и составление ведется именно при этих условиях, всегда указываемых в таблицах. При таких же условиях проводится и анализ, и по осуществлению той или иной гомологической пары находится процентное содержание. Имеется несколько модификаций метода гомологических пар. Из них главнейшим является метод вспомогательного спектра , применяемый в том случае, когда элементы А и В не обладают достаточным количеством линий. В этом случае линии спектра элемента А определенным образом связываются с линиями другого, более пригодного элемента G, и роль А начинает играть элемент G. Метод гомологических пар разработан Герляхом и Швейтцером. Он применим как к сплавам, так и к растворам. Его точность в среднем около ±10%.
В . Методы с применением фотометрии . 1) Метод Барратта . Фиг. 16 дает представление о методе.

F 1 и F 2 - два искровых промежутка, при помощи которых одновременно возбуждаются спектры стандартного и анализируемого вещества. Свет проходит через 2 вращающихся сектора S 1 и S 2 и при помощи призмы D образует спектры, которые расположены один над другим. Путем подбора вырезок секторов линии исследуемого элемента получают одинаковую интенсивность; концентрация определяемого элемента вычисляется из соотношения величин вырезок. 2) является аналогичным, но с одним искровым промежутком (фиг. 17).

Свет от F разделяется на два пучка и проходит через секторы S 1 и S 2 , при помощи ромба Гюфнера R две полоски спектра получаются одна над другой; Sp - щель спектрографа. Вырезки секторов изменяются до получения равенства интенсивности линии примеси и какой-либо близлежащей линии основного вещества и по соотношению величин вырезок высчитывается %-ное содержание определяемого элемента. 3) При применении в качестве фотометра вращающегося логарифмического сектора линии получают на спектрограммах клинообразный вид. Один из таких секторов и его положение относительно спектрографа при работе изображены на фиг. 18, а и б.

Вырезка сектора подчиняется уравнению
- lg Ɵ = 0,3 + 0,2l
где Ɵ - длина дуги в частях полной окружности, находящаяся на расстоянии I, измеренном в мм по радиусу от его конца. Мерой интенсивности линий является их длина, т. к. с изменением концентрации элемента длина его клинообразных линий также изменяется. Предварительно по образцам с известным содержанием строится диаграмма зависимости длины какой-либо линии от %-ного содержания; при анализе на спектрограмме измеряется длина той же линии и по диаграмме находится процентное содержание. Имеется несколько различных модификаций этого метода. Следует указать на модификацию Шейбе, применявшего т. н. двойной логарифмический сектор. Вид этого сектора изображен на фиг. 19.

Линии исследуются затем при помощи специального аппарата. Точность, достижимая при помощи логарифмических секторов, ±(10-15)%; модификация Шейбе дает точность ±(5-7)%. 4) Довольно часто применяется фотометрирование спектральных линий при помощи свето- и термоэлектрических спектрофотометров самых различных конструкций. Удобными являются термоэлектрические фотометры, выработанные специально для целей количественного анализа. Для примера на фиг. 20 приведена схема фотометра по Шейбе:

L– постоянный источник света с конденсором К, М – фотографическая пластинка с исследуемым спектром, Sp - щель, О 1 и О 2 - объективы, V - затвор, Th - термоэлемент, который присоединяется к гальванометру. Мерой интенсивности линий является отклонение стрелки гальванометра. Реже пользуются саморегистрирующими гальванометрами, дающими запись интенсивности линий в виде кривой. Точность анализа при применении этого типа фотометрии составляет ±(5-10)%. При сочетании с другими методами количественного анализа точность м. б. повышена; так, например, метод трех линий Шейбе и Шнеттлера, являющийся сочетанием метода гомологических пар и фотометрических измерений, в благоприятных случаях может дать точность ±(1-2)%.
Спектральные методы анализа основаны на изучении оптических спектров испускания или поглощения. Различают атомно-абсорбционный метод спектрального анализа (анализ по спектрам поглощения) и эмиссионный спектральный анализ (анализ по спектрам испускания). Спектральный анализ широко применяют для качественного и количественного анализа различных веществ. По характеристическим линиям спектра можно определять элементный состав вещества, а интенсивность спектральной линии является мерой концентрации вещества в пробе.
Эмиссионная спектроскопия
Атомы элементов в возбужденном состоянии испускают излучение со строго определенной длиной волны. Спектры испускания (эмиссионные спектры) для каждого элемента индивидуальны, они состоят из определенного набора характерных линий, по которым можно определять элементный состав вещества и его концентрацию.
При эмиссионном спектральном анализе исследуемую пробу испаряют или сжигают, если это жидкое или твердое вещество, затем подвергают действию высокой температуры или электрического заряда для перевода атомов в возбужденное состояние и регистрируют спектр. Качественный эмиссионный анализ сводится к расшифровке линий в спектре анализируемого образца. Количественный анализ основан на сравнении интенсивности спектральных линий образца с интенсивностью линий в спектре стандартного образца, содержание определяемого элемента в котором известно.
Источниками возбуждения могут служить пламя, электрическая дуга, искра, импульсный или электровакуумный разряд. Дуговой разряд дает температуру 5000-7000 °С, при которой в возбужденное состояние переходят атомы большинства элементов. В высоковольтной искре с температурой 7000-15000 °С возбуждаются атомы элементов с высоким потенциалом возбуждения. Импульсный и электровакуумные разряды используют для возбуждения инертных газов.
По методу регистрации спектра различают несколько видов эмиссионного спектрального анализа. При визуальном анализе качественный состав определяют непосредственным наблюдением видимого спектра. Более точен фотографический анализ, по которому спектр фотографируют на фотопластинку, которую затем рассматривают на спектропроекторе при качественных определениях или фотометрируют с помощью микрофотометра при количественных определениях. На фотографической пластинке получают фиксированный ряд линий, соответствующих спектральным линиям исследуемого образца, степень почернения которых пропорциональна интенсивности этих линий.
Для расшифровки спектрограмм используют спектропроекторы. Отечественной промышленностью выпускается спектропроектор ПС-18, который дает возможность получить на экране увеличенные в 20 раз небольшие участки спектра, облегчая их расшифровку при экспрессном качественном или полуколичественном анализе.
Плотность почернения линий на фотопластинке измеряют с помощью микрофотометров. Световой поток пропускают через незачерненную часть фотопластинки, а затем направляют его на фотоэлемент с гальванометром. Отмечают отклонение стрелки гальванометра по шкале. Затем световой поток пропускают через зачерненную часть пластинки и снова отмечают отклонение стрелки гальванометра. Плотность почернения определяют по уравнению:
где I0 - интенсивность света, прошедшего через незачерненную часть фотопластинки; I - интенсивность света, прошедшего через зачерненную часть фотопластинки.
Поскольку плотность почернения пропорциональна концентрации элемента, по показаниям гальванометра строят градуировочный график зависимости почернения от концентрации. По такому графику затем определяют содержание элемента. Для определения плотности почернения линий на спектрограмме применяют микрофотометр МФ-2 (или МФ-4) и двухлучевой микрофотометр ИФО-451.
При фотоэлектрическом эмиссионном анализе аналитические линии регистрируют с помощью фотоэлементов. Результат анализа указывается на шкале измерительного прибора или фиксируется на ленте самозаписывающего прибора.
Кварцевый спектрограф ИСП-28. Спектрограф ИСП-28 используют для получения спектров в интервале длин волн 200-600 нм. На нем проводят качественный и количественный анализы металлов, сплавов, руд, минералов и других материалов. На рис. 126 показана оптическая схема прибора. Свет от источника 1 (дуга или искра) через трехлинзовый конденсор 3-5, защищенный от брызг металлов кварцевой пластинкой 2, направляется в щель 6, находящуюся в фокусе зеркального объектива 8. Отраженный от этого объектива параллельный пучок света направляется на кварцевую призму 9. Подвергшийся дисперсии свет кварцевым объективом 10 фокусируется на эмульсии фотопластинки 11.

Другие спектрографы. Кварцевый лабораторный спектрограф ИСП-30 настольного типа применяется для качественного анализа металлов, сплавов и руд; стеклянный трехпризменный спектрограф ИСП-51 используется для анализа веществ, содержащих элементы с малым числом спектральных линий. Для анализа веществ, содержащих элементы с особо сложными спектрами, используют спектрограф СТЭ-1. Для качественного и количественного анализа металлов, руд, минералов и др. применяют длиннофокусный спектрограф ДФС-8 (три модификации) с дифракционными решетками и дифракционный спектрограф ДФС-452.
Пламенная фотометрия
Пламенная фотометрия является одним из наиболее точных методов эмиссионного спектрального анализа. Этот метод широко применяют для определения щелочных и щелочноземельных металлов. Сущность метода пламенной фотометрии заключается в следующем.
Раствор анализируемого вещества сжатым воздухом разбрызгивается в зону пламени газовой горелки, в которой сгорают ацетилен, водород, светильный или какой-либо другой газ. Пламя горелки служит также источником энергии для возбуждения атомов. Оптическое устройство выделяет спектральную линию определяемого элемента и измеряет ее интенсивность с помощью фотоэлемента. Интенсивность спектральной линии пропорциональна концентрации соли в растворе (в определенных границах). Концентрацию элемента определяют по градуировочному графику. Ниже приведены состав некоторых горючих газовых смесей и средняя температура, получаемая при их сжигании (в °С):

Портативный пламенный фотометр ППФ-УНИЗ. Принципиальная схема фотометра ППФ-УНИЗ представлена на рис. 127. Горючий газ из баллона (или городской сети) проходит через маностат 2, буферную бутыль 3, фильтр 4 и поступает через микрокран 5 в смеситель 7, выполняющий одновременно функцию каплеуловителя. Давление газа после маностата поддерживается постоянным с помощью микрокрана 5 и измеряется U-образным жидкостным манометром 6. Избыток газа выходит в лабораторную горелку 1 и сжигается.

Сжатый воздух из компрессора (без применения масляной смазки) или из баллона поступает в буферную бутыль 3", затем в фильтр 13. Давление воздуха поддерживается постоянным с помощью микрокрана 12 и измеряется манометром 11. Воздух поступает в распылитель 8, куда засасывается анализируемый раствор из стакана 10. Раствор в виде мелкораспыленного аэрозоля поступает в смеситель 7, где смешивается с горючим газом. Выходящая из смесителя газовоздушная смесь, содержащая в распыленном состоянии исследуемый элемент, через каплеуловитель 14 поступает в горелку 20.
Длина волны желтой линии пламени натрия составляет 589±5 мкм, красной линии кальция - 615±5 мкм, инфракрасной линии калия - 766±5 мкм. Интенсивность этих линий фиксируют фотоэлементом 16, снабженным сменными интерференционными светофильтрами 17 и диафрагмами 18. При определении натрия и кальция используют селеновые фотоэлементы типа АФИ-5 с чувствительностью 460-500 мкА/лм, для определения калия - сернисто-серебряный фотоэлемент типа ФЭСС-УЗ с чувствительностью 6000-9000 мкА/лм. Фотоэлементы и светофильтры защищены от прямого теплового излучения пламени стеклянным экраном 19. Возникающие фототоки регистрируются магнитоэлектрическим микроамперметром 21 типа М-95, к которому два из трех фотоэлементов присоединены по компенсационной схеме через электрический переключатель 15.
Перед началом работы с прибором открывают дверку 10 (рис. 128) и закрепляют ее с помощью фиксатора. К сливной трубке 14 распылителя 12 подсоединяют резиновую трубку и опускают ее в сосуд с запорной жидкостью высотой 20-25 см. Под всасывающую трубку 13 распылителя подставляют стакан вместимостью 25-30 мл с дистиллированной водой. На дверку устанавливают защитное устройство (козырек) 11 и включают прибор в сеть переменного тока в 220 В (50 Гц). Включают компрессор для подачи воздуха и, медленно вращая рукоятку микрокрана «воздух» 4 против часовой стрелки, добиваются хорошего распыления дистиллированной воды, т.е. образования высокодисперсного аэрозоля. Оптимальное давление воздуха (4-8)*10000 Па (0,4-0,8 атм) не должно изменяться в течение всего времени измерения.

Медленно вращая рукоятку микрокрана «газ» 5, подают газ в горелку и через 10-20 с зажигают его у входа в горелку и на выходе из маностата. Подачу газа регулируют так, чтобы внутренний конус пламени окрашивался в зеленый цвет, а внешний - в голубовато-синий. С помощью рукоятки 9 устанавливают горелку в таком положении, при котором внутренний конус пламени опущен на 5-6 см ниже кромки входного отверстия диафрагмы.
Измерения начинают после 20-минутного прогревания фотометрической ячейки. В период прогревания диафрагма ячейки должна быть полностью открыта, микроамперметр включают на низкую чувствительность (1,0 мкА) и в пламя горелки вводят дистиллированную воду. После прогревания фотоэлектрической ячейки диафрагму закрывают, рукоятку микроамперметра 6 переключают на высшую чувствительность (0,1 мкА) и указатель микроамперметра устанавливают на нуль, вращая головку корректора, находящуюся на правой боковой стороне прибора.
Для построения градуировочного графика готовят серию стандартных растворов. Для приготовления исходного раствора 2,385 г хлорида калия KCl (хч) растворяют в мерной колбе вместимостью 500 мл и разбавляют водой до метки. Отбирают пипеткой 5,00 мл этого раствора в мерную колбу вместимостью 500 мл и разбавляют дистиллированной водой до метки (разбавление в 100 раз). Полученный раствор содержит 25 мг калия в 1 мл, из него готовят растворы, содержащие 5, 10, 15 и 20 мг калия в 1 мл. Для этого в мерные колбы вместимостью 100 мл отбирают пипеткой 20, 40, 60 и 80 мл раствора с содержанием калия 25 мг/мл и разбавляют объем водой до метки.
Эти растворы последовательно вводят в пламя горелки и записывают показания микроамперметра. При переходе от одного раствора к другому распылитель промывают дистиллированной водой до возвращения стрелки микроамперметра к нулю. По полученным данным строят градуировочный график: показания микроамперметра (по оси абсцисс) - концентрация определяемого элемента (по оси ординат) (в мг/мл).
Для определения концентрации элемента в исследуемом растворе его вводят в пламя горелки и записывают показания микроамперметра, по которым, пользуясь градуировочным графиком, находят концентрацию определяемого элемента. В течение всего процесса анализа необходимо поддерживать постоянство давления воздуха и газа.
Кроме метода определения концентрации по градуировочному графику применяют метод ограничивающих растворов, т.е. снимают показания микроамперметра при анализе исследуемого раствора и параллельно показания прибора при анализе стандартных: растворов с меньшей и большей концентрацией. Содержание калия (в мг/л) вычисляют по формуле
![]()
где c1 - содержание калия в более концентрированном стандартном растворе; c2 - содержание калия в менее концентрированном стандартном растворе; I1 - показания микроамперметра при анализе стандартного раствора с большей концентрацией; I2 - показания микроамперметра при анализе стандартного раствора с меньшей концентрацией; Ix - показания микроамперметра при анализе исследуемого раствора.
Пламенный фотометр Flapho-4. Двухканальный прибор для серийного определения содержания натрия, калия, кальция, лития и свинца с высокой чувствительностью. Выпускается в ГДР.
Исследуемый раствор пробы всасывается протекающим через; распылитель сжатым воздухом и превращается в аэрозоль. Аэрозоль поступает в специальный резервуар, где к нему примешивается горючий газ (ацетилен или пропан), и полученная смесь подводится к горелке, окруженной очищенным воздухом. В газовом пламени исследуемое вещество испаряется, и его атомы возбуждаются. Металлизированный интерференционный фильтр выделяет из общего спектра пламени монохроматический компонент излучения, который попадает на селеновый фотоэлемент. Образующийся прерывистый фототок усиливается и подводится к измерительному или регистрирующему прибору. Схема прибора представлена на рис. 129.

Другие пламенные фотометры: фотометр пламенный ФП-101 трехканальный для определения концентрации Na, K, Ca и Li; фотометр пламенный ПФМ для количественного определения концентраций щелочных и щелочноземельных элементов, а также магния, бора, хрома и марганца; пламенно-фотометрические анализаторы жидкости ПАЖ-1 и БИАН-140 для определения микроколичеств K, Na, Ca и Li в растворах, фотометр пламенный для определения Na и K в биологических жидкостях.
Атомно-абсорбционная спектрофотометрия
Свободные атомы в невозбужденном состоянии, находящиеся в зоне низкотемпературного пламени, обладают способностью избирательно поглощать свет. Длина волны света, поглощаемого атомами элемента, совпадает с длиной волны света, испускаемого атомами этого элемента. Следовательно, по характеристическим линиям спектра поглощения и их интенсивности можно проводить анализ веществ, определяя их состав и концентрацию составляющих его элементов.
Для проведения атомно-абсорбционного анализа исследуемое вещество испаряют, подавая его в зону низкотемпературного пламени. Молекулы испарившегося вещества диссоциируют на атомы. Поток света, в спектре которого имеется линия света, поглощаемая веществом, пройдя через это пламя, ослабляется, и тем больше, чем выше концентрация анализируемого вещества.
На рис. 130 представлена принципиальная схема установки для атомно-абсорбционного анализа. Свет от разрядной трубки 1 (полый катод) проходит через пламя горелки 2 и фокусируется на щели монохроматора 3. Затем излучение попадает на фотоумножитель, или фотоэлемент 4. Монохроматор выделяет из общего светового потока излучение с длиной волны, поглощаемой исследуемым элементом. Ток усиливается в блоке 5 и регистрируется измерительным устройством 6.

Определение заключается в измерении отношения интенсивностей света, прошедшего через пламя с введенным в него анализируемым веществом и без него. Поскольку интенсивность спектральной линии исследуемого элемента в пламени горелки оказывается больше, чем их интенсивность излучения от полого катода, излучение последнего модулируют. Модуляция излучения (изменение амплитуды и частоты колебаний) осуществляется с помощью вращающегося диска с отверстиями (модулятор 7), расположенного между полым катодом и пламенем. Усилитель 5 должен иметь максимальный коэффициент усиления для той же частоты, с которой модулируется излучение полого катода.
Атомно-абсорбционный спектрофотометр AAS-1. Предназначается для абсорбционного и эмиссионного спектрального анализа. Дает возможность определять 65 элементов.
Принцип действия. Жидкая проба распыляется с помощью газа-окислителя, смешивается с горючим газом (ацетилен или пропан) и сжигается в пламени горелки. Через пламя горелки проходит излучение от лампы с полым катодом. После выделения дифракционным монохроматором подходящей линии излучение направляется на фотоумножитель. Постоянная составляющая тока, вызванная собственным излучением, подавляется. Сигнал от фото-умножителя усиливается, выпрямляется чувствительным выпрямителем и регистрируется. Прибор настраивается и контролируется по стандартным растворам.
На рис. 131 приведена схема атомно-абсорбционного спектрофотометра AAS-1.

Устройство прибора. Прибор имеет арматурный комплекс для снабжения газами, систему распыления и сжигания, сменное устройство для ламп с полыми катодами, оптическую систему я приемное устройство с усилителем и индикатором.
Пламя горелки питается смесью ацетилена или пропана и сжатого воздуха. Газы поступают в систему сжигания из обычных баллонов с отрегулированными (первичными) редукторами давления. Подача воздуха, свободного от масла, обеспечивается мембранным компрессором (16 л/мин под давлением 3*100000 Па (3 атм)). Арматурный комплекс прибора имеет регулируемые (вторичные) редукторы и расходомеры для контроля расхода каждого газа, а также керамические спеченные пылевые фильтры и склянку для дополнительного промывания ацетилена. Предохранительный клапан автоматически прекращает доступ горючего газа при снижении рабочего давления сжатого воздуха (например, вследствие перегиба или отрыва подводящего шланга); клапан исключает неправильный порядок подачи газов при зажигании пламени.
Система распыления и сжигания находится за съемным окном из многослойного стекла, позволяющего наблюдать за работой системы. Распылитель с кольцевым соплом обладает большим коэффициентом распыления и характеризуется низким расходом жидкости (3,4 мл/мин, или 0,5 мл за время всего анализа). Горелка оснащена сменными головками-насадками - одной щелевой для абсорбционного анализа (рис. 132, а) и двумя многодырчатыми (горелками Мекера с сеткой) для эмиссионного анализа (рис. 132,6).

Юстируемые держатели для четырех ламп с полыми катодами находятся в устройстве, позволяющем осуществлять быструю смену ламп. После замены одной из ламп держатели в юстировке не нуждаются.
Оптическая система направляет излучение лампы в виде узкого пучка на пламя. За счет бокового смещения тубуса с изображающей системой добиваются однократного или трехкратного прохождения излучения через пламя для повышения чувствительности анализа. Светосильный дифракционный монохроматор выделяет из линейчатого спектра данной лампы с полым катодом желаемую резонансную линию. Ширину щели монохроматора регулируют в пределах от 0 до 2 мм.
Прецизионная дифракционная решетка с 1300 штрихами на 1 мм и угловой дисперсией 1,5 нм/мм обладает большой разрешающей способностью. Спектральный интервал решетки от 190 до 820 нм.
Приемником излучения служит 12-каскадный фотоумножитель. Измерительный усилитель, блок питания ламп с полым катодом и фотоумножители работают на транзисторах и способны компенсировать колебания напряжения сети от +10 до -15%.
Показания прибора отсчитывают по стрелочному индикатору, имеющему три шкалы: логарифмическая шкала коэффициента погашения от 0 до 1,5; линейная шкала от 0 до 100 и шкала рабочих напряжений от 0 до 16 мВ. К прибору может быть подключено регистрирующее или вычислительное устройство для определения концентрации или для обработки данных. Чувствительность определений (в мг/л) составляет:
![]()
Прибор работает от сети переменного тока 220 В, 50 Гц. Выпускается в ГДР.
Другие отечественные атомно-абсорбционные спектрофотометры: атомно-абсорбционный спектрофотометр С-302 для определения микроколичеств железа, меди, цинка, кобальта, никеля, висмута, кальция и других элементов; автоматизированный атомно-абсорбционный спектрофотометр АА-А для определения кальция и меди с повышенной чувствительностью; «Сатурн» - пламенный атомно-абсорбционный полуавтоматический регистрирующий спектрофотометр для определения 32 элементов; «Спектр-1» - атомно-абсорбционный спектрофотометр для экспрессного определения более 40 элементов чувствительностью примерно 0,2 мкг/мл.
В Англии выпускается атомно-абсорбционный спектрофотометр Перкин-Эльмер, модель 603. Прибор построен по двухлучевой схеме, скомбинирован с микрокомпьютером. Обеспечивает высокую точность и экспрессность определения. Для зажигания пламени используется горючая смесь кислород-ацетилен.
Спектры, способы их получения, особенности, классификация и использование для аналитических целей. Основные элементы спектральных приборов и их назначение
Спектральные методы анализа - это методы, основанные на определении химического состава и строения веществ по их спектру.
Спектром вещества называют упорядоченное по длинам волн электромагнитное излучение, испускаемое, поглощаемое, рассеиваемое или преломляемое веществом. Методы, основанные на получении и изучении спектров испускания (эмиссии) электромагнитного излучения (энергии), называют эмиссионными, поглощения (абсорбции) - абсорбционными, рассеяния - методами рассеяния, преломления - рефракционными.
Спектр вещества получают, воздействуя на него температурой, потоком электронов, световым потоком (электромагнитной энергией) с определённой длиной волны (частоты излучения) и другими способами. При определённой величине энергии воздействия вещество способно перейти в возбуждённое состояние. При этом происходят процессы, приводящие к появлению в спектре излучения с определённой длиной волны (табл.2.2.1).
Излучение, поглощение, рассеяние или рефракция электромагнитного излучения может рассматриваться как аналитический сигнал, несущий информацию о качественном и количественном составе вещества или о его структуре. Частота (длина волны) излучения определяется составом исследуемого вещества, а интенсивность излучения пропорциональна числу частиц, вызвавших его появление, т.е. количеству вещества или компонента смеси.
Каждый из аналитических методов обычно использует не полный спектр вещества, охватывающий диапазон длин волн от рентгеновских излучений до радиоволн, а только определённую его часть. Спектральные методы обычно различают по диапазону длин волн спектра, являющемуся рабочим для данного метода: ультрафиолетовые (УФ), рентгеновские, инфракрасные (ИК), микроволновые и т.д.
Методы, работающие в УФ, видимом и ИК диапазоне называют оптическими. Они больше всего применяются в спектральных методах вследствие сравнительной простоты оборудования для получения и регистрации спектра.
Спектры оптического диапазона являются результатом изменения энергии атомов или молекулах.
Таблица 2.2.1
| Вид излучения | Атомные и молекулярные процессы | Источники возбуждения | Детекторы излучения | |
| , нм | название | |||
| 10-3 | -излучение | Ядерные | Циклотроны | Счётчики Гейгера, |
| 10-2 | Рентгеновское | реакции | сцинциляционные счётчики, фотопластины | |
| 10-1 | Переходы внешних | Рентгеновские трубочки | ||
| 100 | электронов | |||
| 101 | УФ ваккумное | |||
| 2·102 | УФ дальнее | Переходы внешних электронов | Рентгеновские трубочки, искра, пламя, дуга | Фотоэлементы, фотоматериалы |
| 3·102 | УФ ближнее | |||
| 375-750 | Видимое | Глаз, фотоэлемент | ||
| 104 | ИК ближнее | Колебания молекул | Нагретые металлические нити | Вакуумные термопары, |
| 105 | Дальнее | Вращение молекул | боллометры | |
В результате изменения энергии атома или молекулы они переходят из основного состояния с минимально возможной внутренней энергией Е0 в возбужденное состояние с энергией Е1. Внутренняя энергия является величиной дискретной (квантовой), поэтому переход атома или молекулы из основного состояния в другое всегда сопровождается скачкообразным изменением энергии, т.е. получением или отдачей порции (кванта) энергии.
Квантами электромагнитного излучения являются фотоны, энергия которых связана с частотой и длиной волны излучения известным соотношением
Е = h · =
,где Е = Е1 - Е2, Е1 - энергия начального, а Е2 - энергия конечного состояния атома или молекулы, между которыми происходит переход; h - постоянная Планка; с - скорость света; - частота; - длина волны электромагнитного излучения.
При возбуждении атома происходит перемещение электронов с внешних заполненных уровней на незаполненные более высокие энергетические уровни.
В возбуждённом состоянии атом не может долго находиться. Он стремится отдать полученную избыточную энергию и возвратиться в невозбуждённое состояние. Через очень короткое время (10-8 - 10-7с) атом самопроизвольно возвращается из возбуждённого состояния в основное или промежуточное.
При переходе электрона с верхнего уровня на нижний выделяется фотон - квант излучения с определёнными и .
Схематично электронные переходы в атомах между различными состояниями, сопровождающиеся испусканием и поглощением квантов электромагнитного излучения, можно представить в виде схемы (рис.2.2.1).

Горизонтальными линиями на рис.2.2.1. изображены уровни энергии различных состояний атома. Уровень Е0 это уровень основного состояния; Е1, Е2, Е3 - уровни возбуждённых состояний в порядке возрастания их энергии. Вертикальные стрелки соответствуют испусканию (стрелка вниз) или поглощению () фотона. Очевидно, что
01 = 10, 13 = 31 и т.д.
Совокупность фотонов, испускаемых или поглощаемых при каком - либо одном электронном переходе атома, создающая излучение с одной длиной волны, называется спектральной линией. Длина волны спектральной линии может быть определена из соотношения =
. Совокупность спектральных линий, относящихся к определённому атому (молекуле), образует спектр данного атома (молекулы).Спектр, обусловленный переходом при Е1 Е2, называется спектром испускания, а при Е1 Е2 - спектром поглощения. Переходы и соответствующие спектральные линии, проходящие с основного энергетического уровня или на него, называются резонансными.
Для возбуждения спектральной линии необходима определённая энергия, называемая потенциалом возбуждения. Если сообщить атому слишком большую энергию, то может произойти полное удаление электрона, т.е. ионизация атома. Необходимая для этого энергия называется потенциалом ионизации. Резонансные линии самые яркие и характеризуются наименьшим потенциалом возбуждения.
Изменение энергии молекулы сопровождается изменением как энергии колебаний и вращений, т.е. у молекулы нет чисто электронных переходов, а возможны только электронно-колебательно-вращатель-ные (ЭКВ) переходы. Число возможных ЭКВ переходов у молекулы значительно больше, чем у атомов, поэтому, как правило, спектры молекул сложнее и состоят из большего числа спектральных линий в оптическом диапазоне длин волн. Принципиальную схему энергетических уровней молекулы можно представить следующим образом (рис.2.2.2).

Рис.2.2.2. Схема энергетических уровней молекулы
Как для молекул, так и для атомов проявляются не все мыслимые переходы. Переходы регламентируются так называемыми правилами отбора: разрешенными являются переходы, при которых квантовое число меняется на единицу (например, Sp, pd и т.д.).
Для аналитических целей можно использовать как эмиссионные, так и абсорбционные спектры, поскольку они взаимосвязаны. Например, свет, излучаемый раскалёнными парами металлического натрия, пропущенный через призму, даёт две очень близкие желтые линии с длинами волн 589,0 и 589,6 мкм. Это так называемые D - линии натрия. С другой стороны, если пропускать полихроматический белый свет (т.е. Совокупность пучков света со всеми длинами волн) через пары натрия, а затем разложить его на составляющие цвета в стеклянной призме, то на фоне непрерывного спектра будут обнаружены две чёрные линии как раз на месте D - линий. Следовательно, пары натрия поглощают излучение именно с теми длинами волн, какие они испускают при возбуждении.
Это - общая закономерность, поэтому спектральный анализ можно проводить как по спектру испускания, так и по спектру поглощения. Первый способ удобен для анализа материалов, в которых легко возбуждается спектр испускания составляющих веществ, например металлов и газов, а второй - более удобен при анализе материалов, в которых трудно вызвать возбуждение составляющих веществ (например, растворы).
Эмиссионные спектры делятся на сплошные, полосатые, линейчатые (рис.2.2.3). Сплошные (или непрерывные) спектры содержат все длины волн в определённом интервале.
Их испускают раскалённые которые находятся на таких расстояниях друг от друга, что их излучение можно считать независимым. Газы и пары металлов имеют линейчатые спектры.
Линии в спектрах атомов расположены не беспорядочно, а объединяются в группы, называемые сериями. Расстояния между линиями в серии закономерно убывают по мере перехода от более длинных волн к более коротким.
Бальмеером для простейшего линейчатого спектра водорода было обнаружено, что частоты спектральных линий в сериях, расположенных в различных областях электромагнитного излучения, находятся в определённой закономерной связи друг с другом, которую в общем виде для всех элементов выразили зависимостью

или в определённых случаях
Спектральный анализ подразделяют на несколько самостоятельных методов. Среди них выделяют: инфракрасную и ультрафиолетовую спектроскопию, атомно-абсорбционный, люминесцентный и флуоресцентный анализ, спектроскопию отражения и комбинационного рассеяния, спектрофотометрию, рентгеновскую спектроскопию, а также ряд других методов.
Абсорбционный спектральный анализ основан на изучении спектров поглощения электромагнитного излучения. Эмиссионный спектральный анализ проводится по спектрам испускания атомов, молекул или ионов, возбужденных различными способами.
Атомно-эмиссионный спектральный анализ
Спектральным анализом часто называют только атомно-эмиссионный спектральный анализ, который основан на исследовании спектров испускания свободных атомов и ионов в газовой фазе. Его проводят в области длин волн 150-800 нм. В источник излучения вводят пробу исследуемого вещества, после чего в нем происходит испарение и диссоциация молекул, а также возбуждение образовавшихся ионов. Они испускают излучение, которое фиксируется регистрирующим устройством спектрального прибора.
Работа со спектрами
Спектры проб сравнивают со спектрами известных элементов, которые можно найти в соответствующих таблицах спектральных линий. Так узнают состав анализируемого вещества. Количественный анализ подразумевает концентрации данного элемента в анализируемого веществе. Ее узнают по величине сигнала, например, по степени почернения или оптической плотности линий на фотопластинке, по интенсивности светового потока на фотоэлектрическом приемнике.
Виды спектров
Непрерывный спектр излучения дают вещества, находящиеся в твердом или жидком состоянии, а также плотные газы. В таком спектре нет разрывов, в нем представлены волны всех длин. Его характер зависит не только от свойств отдельных атомов, но и от их взаимодействия друг с другом.
Линейчатый спектр излучения характерен для веществ в газообразном состоянии, при этом атомы почти не взаимодействуют друг с другом. Дело в том, что изолированные атомы одного химического элемента излучают волны строго определенной длины волны.
При увеличении плотности газа спектральные линии начинают расширяться. Для наблюдения такого спектра используют свечение газового разряда в трубке или паров вещества в пламени. Если пропускать белый свет через неизлучающий газ, на фоне непрерывного спектра источника появятся темные линии спектра поглощения. Газ интенсивнее всего поглощает свет тех длин волн, которые он испускает в нагретом состоянии.
Одним из основных методов анализа химического состава вещества является спектральный анализ. Анализ его состава производится, на основании изучения его спектра. Спектральный анализ — используется в различных исследованиях. С его помощью открыт комплекс химических элементов: Не, Ga, Cs. в атмосфере Солнца. А также Rb, Inи XI, определён состав Солнца и большинства других небесных тел.
Отрасли применения
Спектральная экспертиза, распространена в:
- Металлургии;
- Геологии;
- Химии;
- Минералогии;
- Астрофизике;
- Биологии;
- медицине и др.
Позволяет находить в изучаемых объектах малейшие количества устанавливаемого вещества (до 10 — MS) Спектральный анализ делится на качественный и количественный.
Методы
Способ установления химического состава вещества на основе спектра – это и есть основа спектрального анализа. Линейчатые спектры обладают неповторимой индивидуальностью, так же как и отпечатки пальцев у людей, или же узор снежинок. Неповторимость рисунков на коже пальца – это большое преимущество для розыска преступника. Поэтому благодаря особенности каждого спектра имеется — возможность установить химическое содержание тела, проведя анализ химического состава вещества. Даже если его масса элемента не превышает 10 — 10 г, с помощью спектрального анализа его можно обнаружить в составе сложного вещества. Это достаточно чувствительный метод.
Эмиссионный спектральный анализ
Эмиссионный спектральный анализ — это ряд методов установления химического состава вещества по его эмиссионному спектру. В основу способа установления химического состава вещества – спектральной экспертизы, положены закономерности в спектрах испускания и спектрах поглощения. Данный метод позволяет выявить миллионные доли миллиграмма вещества.
Существуют методы качественной и количественной экспертизы, в соответствии с установлением аналитической химии как предмета, целью которой является формирование способов установления химического состава вещества. Методы идентификации вещества, становятся крайне важными в пределах качественного органического анализа.
По линейчатому спектру паров какого-либо из веществ есть возможность установить, какие химические элементы содержаться в его составе, т.к. любой химический элемент имеет личный специфический спектр излучения. Подобный метод установления химического состава вещества именуется качественным спектральным анализом.
Рентгеноспектральный анализ
Существует еще один метод определения химического вещества, называемый рентгеноспектральным анализом. Рентгеноспектральный анализ основан на активации атомов вещества при облучении его рентгеновскими лучами, процесс называется вторичным или флуоресцентным. А также возможна активация при облучении электронами больших энергий, в этом случае процесс именуют прямым возбуждением. В результате перемещения электронов в более глубоких внутренних электронных слоях появляются линии рентгеновского излучения.
Формула Вульфа — Брэггов позволяет устанавливать длины волн, в составе рентгеновского излучения, при применении кристалла популярной структуры с известным расстоянием d. Это и есть основание метода определения. Изучаемое вещество бомбят стремительными электронами. Помещают его, к примеру, на анод разборной рентгеновской трубки, впоследствии чего оно источает характерные рентгеновские лучи, которые падают на кристалл известной структуры. Измеряют углы и рассчитывают по формуле соответствующие длины волн, после фотографирования возникающей при этом дифракционной картине.
Приемы
В настоящее время все методы химического анализа основаны на двух приемах. Либо на: физическом приеме, либо на химическом приеме сравнения устанавливаемой концентрации с ее единицей измерения:
Физический
Физический приём основан на способе соотнесения с эталоном единицы величины количества компонента путем измерения его физического свойства, который зависит от его содержания в пробе вещества. Пробно определяют функциональную зависимость «Насыщенность свойства – содержание компонента в пробе» способом градуировки средства измерения данного физического свойства по устанавливаемому компоненту. Из градуировочного графика получают количественные отношения, построенного в координатах: «насыщенность физического свойства — концентрация устанавливаемого компонента».
Химический
Химический приём используется в способе соотнесения с эталоном единицы величины количества компонента. Тут используются законы сохранения количества или массы компонента при химических взаимодействиях. На химических свойствах химических соединений, основаны химические взаимодействия. В пробе вещества осуществляют химическую реакцию, отвечающую поставленным требованиям, для определения искомого компонента, и производится замер объёма или массы, принимающих участие в конкретной химической реакции компонентов. Получают количественные отношения, далее записывается количества эквивалентов компонента для данной химической реакции или закон сохранения массы.
Приборы
Приборами для анализа физико-химического состава вещества являются:
- Газоанализаторы;
- Сигнализаторы предельно допустимых и до взрывоопасных концентраций паров и газов;
- Концентратомеры жидких растворов;
- Плотномеры;
- Солемеры;
- Влагомеры и др. схожие по назначению и комплектности приборы.
Со временем все более увеличивается круг анализируемых объектов и повышается скорость и правильность анализа. Одним из главнейших приборных методов установления атомного химического состава вещества становится спектральный анализ.
ПРИМЕЧАНИЕ:
Цена химической экспертизы указана с учетом налогов. Транспортные расходы оплачиваются отдельно.