SPEKTRALANALYS(med emissionsspektra) används i nästan alla sektorer av ekonomin. Används i stor utsträckning inom metallindustrin för snabb analys av järn, stål, gjutjärn, såväl som olika specialstål och färdiga metallprodukter, för att bestämma renheten hos lätta, icke-järnhaltiga och ädla metaller. Spektralanalys används i stor utsträckning inom geokemi när man studerar sammansättningen av mineraler. Inom den kemiska industrin och närliggande industrier används spektralanalys för att bestämma renheten hos tillverkade och använda produkter, för att analysera katalysatorer, olika rester, sediment, grumligheter och tvättvatten; inom medicin - för upptäckten av metaller i olika organiska vävnader. Ett antal specialproblem som är svåra att lösa eller inte kan lösas på annat sätt löses med spektralanalys snabbt och noggrant. Detta inkluderar till exempel fördelningen av metaller i legeringar, studiet av sulfid och andra inneslutningar i legeringar och mineraler; Denna typ av forskning kallas ibland lokal analys.
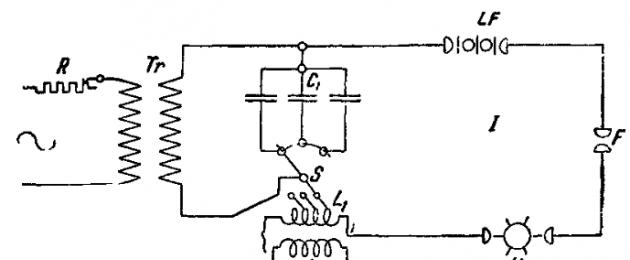
Valet av en eller annan typ av spektralapparat ur synvinkeln av dess dispersions tillräcklighet görs beroende på syftet och målen med spektralanalys. Kvartsspektrografer med större spridning, ger för våglängderna 4000-2200 Ӑ en spektrumremsa som är minst 22 cm lång. För andra element kan det vara Apparater används som ger spektra 7-15 cm långa Spektrografer med glasoptik är i allmänhet av mindre betydelse. Av dessa är kombinerade instrument bekväma (till exempel från Hilger och Fuss företag), som om så önskas kan användas som spektroskop och spektrograf. Följande energikällor används för att erhålla spektra. 1) Flamma av brinnande blandning- väte och syre, en blandning av syre och lysande gas, en blandning av syre och acetylen, eller slutligen luft och acetylen. I det senare fallet når ljuskällans temperatur 2500-3000°C. Lågan är mest lämpad för att erhålla spektra av alkali- och jordalkalimetaller, samt för grundämnen som Cu, Hg och Tl. 2) Voltaisk båge. a) Vanlig, 2 kap. arr. likström, effekt 5-20 A. Den används med stor framgång för kvalitativ analys av svårsmälta mineraler, som införs i bågen i form av bitar eller finmalda pulver. För den kvantitativa analysen av metaller har användningen av en konventionell voltaisk båge en mycket betydande nackdel, nämligen att ytan på de analyserade metallerna täcks med en oxidfilm och att ljusbågens förbränning slutligen blir ojämn. Temperaturen på den voltaiska bågen når 5000-6000°C. b) Intermittent båge (Abreissbogen) av likström med en effekt på 2-5 A vid en spänning på ca 80 V. Med hjälp av en speciell anordning avbryts ljusbågen 4-10 gånger per sekund. Denna exciteringsmetod minskar oxidationen av ytan på de analyserade metallerna. Vid högre spänningar - upp till 220 V och en ström på 1-2 A - kan en intermittent båge också användas för att analysera lösningar. 3) Gnistutsläpp, erhållen med hjälp av en induktionsspole eller, oftare, en lik- eller (helst) växelströmstransformator med en effekt på upp till 1 kW, vilket ger 10 000-30 000 V i sekundärkretsen. Tre typer av urladdningar används, a) Gnisturladdningar utan kapacitans och induktans i sekundärkretsen, kallad ibland med högspänningsbåge (Hochspannungsbogen). Analysen av vätskor och smälta salter med sådana utsläpp är mycket känslig. b) Gnisturladdningar med kapacitans och induktans i sekundärkretsen, ofta även kallad kondenserade gnistor, representerar en mer universell energikälla, lämplig för att excitera spektra av nästan alla element (förutom alkalimetaller), såväl som gaser. Anslutningsschemat visas i fig. 1,

där R är reostaten i primärkretsen, Tr är växelströmstransformatorn, C 1 är kapacitansen i sekundärkretsen I, S är omkopplaren för att ändra induktansen L 1, U är synkronbrytaren, LF är gnistfångaren , F är arbetsgnistgapet. Sekundärkretsen II är avstämd till resonans med sekundärkretsen I med användning av induktans och variabel kapacitans C2; ett tecken på förekomst av resonans är den högsta strömstyrkan som visas av milliammeter A. Syftet med sekundärkretsen II för synkronbrytaren U och gnistfångaren LF är att göra elektriska urladdningar så enhetliga som möjligt både till karaktär och antal över en viss tidsperiod; under normalt arbete införs inte sådana extra anordningar.
När man studerar metaller i sekundärkretsen används en kapacitans på 6000-15000 cm3 och en induktans på upp till 0,05-0,01 N. För att analysera vätskor införs ibland en vattenreostat med ett motstånd på upp till 40000 Ohm i sekundärkretsen . Gaser studeras utan induktans med liten kapacitans. c) Tesla-strömurladdningar, som utförs med den krets som visas i fig. 2,

där V är en voltmeter, A är en amperemeter, T är en transformator, C är en kapacitans, T-T är en Tesla-transformator, F är gnistgapet där det analyserade ämnet införs. Tesla-strömmar används för att studera ämnen som har en låg smältpunkt: olika växt- och organiska preparat, avlagringar på filter etc. Vid spektralanalys av metaller, när det gäller ett stort antal av dem, är de vanligtvis själva elektroder, och de ges någon form, till exempel från de som visas i FIG. 3,

där a är en elektrod gjord av den tjocka tråden som analyseras, b är av tenn, c är en böjd tunn tråd, d är en skiva skuren från en tjock cylindrisk stav, e är en form skuren från stora gjutstycken. Vid kvantitativ analys är det alltid nödvändigt att ha samma form och storlek på elektrodytan exponerad för gnistor. Om mängden metall som analyseras är liten, kan du använda en ram gjord av någon ren metall, till exempel guld och platina, i vilken den analyserade metallen är fixerad, som visas i Fig. 4.
En hel del metoder har föreslagits för att införa lösningar i en ljuskälla. När man arbetar med en låga används en Lundegaard atomizer, schematiskt visad i fig. 5 tillsammans med en speciell brännare.

Luften som blåses genom BC-sprutan fångar upp testvätskan, hälls i en mängd av 3-10 cm 3 i fördjupningen C och för den i form av fint damm till brännare A, där den blandas med gas. För att införa lösningar i bågen, såväl som i gnistan, används rena kol- eller grafitelektroder, på varav en är en fördjupning. Det bör dock noteras att det är mycket svårt att koka kol helt rent. Metoderna som används för rengöring - alternerande kokning i salt- och fluorvätesyra, samt kalcinering i väteatmosfär till 2500-3000 ° C - producerar inte kol fria från föroreningar; Ca, Mg, V, Ti, Al finns kvar (om än spår ), Fe, Si, B. Kol av tillfredsställande renhet erhålls också genom att kalcinera dem i luft med hjälp av en elektrisk ström: en ström på cirka 400 A passerar genom en kolstav med en diameter av 5 mm, och den starka glödlampan som uppnås i detta sätt (upp till 3 000 ° C) är tillräckligt för att inom några sekunder kommer de flesta av föroreningarna som förorenar kolen att avdunsta. Det finns också metoder för att införa lösningar i en gnista, där själva lösningen är den nedre elektroden, och gnistan hoppar till dess yta; en annan elektrod kan vara vilken ren metall som helst. Ett exempel på en sådan anordning visas i fig. 6 Gerlyach flytande elektrod.

Fördjupningen i vilken testlösningen hälls är fodrad med platinafolie eller täckt med ett tjockt lager av guld. I fig. 7 visar en Hitchen-apparat, som även tjänar till att införa lösningar i en gnista.

Från kärl A strömmar testlösningen i en svag ström genom rör B och kvartsmunstycke C in i verkningssfären för gnisturladdningar. Den nedre elektroden, lödd i ett glasrör, fästs vid apparaten med hjälp av ett gummirör E. Fästet C, visat i fig. 7 separat, har ett urtag på ena sidan för murbruk. D - ett säkerhetskärl av glas i vilket ett runt hål görs för utsläpp av ultravioletta strålar. Det är bekvämare att göra denna kärlkvarts utan hål. Toppelektroden F, grafit, kol eller metall, är också försedd med en stänksäker platta. För en "högspänningsbåge" som starkt värmer de analyserade ämnena, använder Gerlach kylda elektroder när han arbetar med lösningar, som visas schematiskt i fig. 8.

En glastratt G fästs på en tjock tråd (6 mm i diameter) med hjälp av en propp K, i vilken isbitar placeras. I den övre änden av tråden är en rund järnelektrod E med en diameter av 4 cm och en höjd av 4 cm fixerad, på vilken en platinakopp P placeras; den senare bör vara lätt avtagbar för rengöring. Den övre elektroden ska också användas. tjock för att undvika smältning. Vid analys av små mängder ämnen - sediment på filter, olika pulver etc. - kan du använda enheten som visas i fig. 9.
En klump tillverkas av testämnet och filterpapper, fuktad för bättre ledningsförmåga med en lösning, till exempel NaCl, placerad på den undre elektroden, ibland bestående av rent kadmium, inneslutet i ett kvartsrör (sämre glas); den övre elektroden är också av ren metall. För samma analyser när man arbetar med Tesla-strömmar används en speciell gnistgapsdesign, som visas i fig. 10 a och b.

I det runda gångjärnet K fixeras en aluminiumplatta E i önskat läge, på vilken en glasplatta G placeras och på den senare - preparat P på filterpapper F. Preparatet fuktas med någon syra- eller saltlösning. Hela detta system är en liten kondensator. För att studera gaser används slutna glas- eller kvartskärl (fig. 11).

För kvantitativ analys av gaser är det bekvämt att använda guld- eller platinaelektroder, vars linjer kan användas för jämförelse. Nästan alla ovan nämnda anordningar för att införa ämnen i en gnista och båge är monterade i speciella stativ under drift. Ett exempel är Gramont-stativet som visas i fig. 12:

med skruv D flyttas elektroderna samtidigt isär och flyttas isär; skruv E används för att flytta den övre elektroden parallellt med den optiska bänken, och skruv C är för lateral rotation av den nedre elektroden; skruv B används för lateral rotation av hela den övre delen av stativet; slutligen, med hjälp av skruv A, kan du höja eller sänka hela den övre delen av stativet; N - står för brännare, glas, etc. Valet av energikälla för ett visst forskningsändamål kan göras med hjälp av följande ungefärliga tabell.

Kvalitativ analys. I kvalitativ spektralanalys beror upptäckten av ett element på många faktorer: arten av det element som bestäms, energikällan, spektralapparatens upplösning, såväl som känsligheten hos fotografiska plattor. När det gäller analysens känslighet kan följande riktlinjer göras. När du arbetar med gnistorladdningar i lösningar kan du öppna 10 -9 -10 -3%, och i metaller 10 -2 -10 -4% av elementet som studeras; vid arbete med en ljusbåge är öppningsgränserna cirka 10 -3 %. Det absoluta belopp som kan vara öppen när man arbetar med en låga, är 10 -4 -10 -7 g, och med gnisturladdningar 10 -6 -10 -8 g av det element som studeras. Den största känsligheten för upptäckt gäller metaller och metalloider - B, P, C; mindre känslighet för metalloider As, Se och Te; halogener, liksom S, O, N i deras föreningar, kan inte användas alls. öppen och m.b. upptäcks endast i vissa fall i gasblandningar.
För kvalitativ analys är de "sista linjerna" av största vikt och i analysen är uppgiften att mest exakt bestämma spektrallinjernas våglängder. I visuella studier mäts våglängder längs spektrometertrumman; dessa mätningar kan endast betraktas som ungefärliga, eftersom noggrannheten vanligtvis är ±(2-З)Ӑ och i Kaiser-tabellerna kan detta felintervall motsvara ca 10 spektrallinjer som tillhör olika element för λ 6000 och 5000Ӑ och ca 20 spektrallinjer för λ ≈ 4000 Ӑ. Våglängden bestäms mycket mer exakt genom spektrografisk analys. I detta fall, på spektrogrammen, med hjälp av ett mätmikroskop, mäts avståndet mellan linjer med en känd våglängd och en bestämd; Hartmanns formel används för att hitta våglängden för den senare. Noggrannheten för sådana mätningar när man arbetar med ett instrument som producerar en spektrumremsa som är cirka 20 cm lång är ± 0,5 Ӑ för λ ≈ 4000 Ӑ, ± 0,2 Ӑ för λ ≈ 3000 Ӑ och ± 0,1 Ӑ för λ ≈ Ӑ. Motsvarande element finns i tabellerna baserat på våglängden. Avståndet mellan linjerna vid normalt arbete mäts med en noggrannhet på 0,05-0,01 mm. Denna teknik kombineras ibland bekvämt med skjutspektra med så kallade Hartmann-luckor, av vilka två typer visas i fig. 13, a och b; Med deras hjälp kan spektrografslitsen göras i olika höjder. Fikon. 13c visar schematiskt fallet med kvalitativ analys av substans X - identifieringen av elementen A och B. Spektrana i FIG. 13, d visar att det i substans Y, förutom element A, vars linjer betecknas med bokstaven G, finns en förorening, vars linjer betecknas z. Med denna teknik kan du i enkla fall utföra en kvalitativ analys utan att behöva mäta avstånden mellan linjerna.

Kvantitativ analys. För kvantitativ spektralanalys är linjer som har största möjliga koncentrationskänslighet dI/dK av största vikt, där I är intensiteten på linjen och K är koncentrationen av det element som ger den. Ju större koncentrationskänslighet desto mer exakt analys. Med tiden har ett antal metoder för kvantitativ spektralanalys utvecklats. Dessa metoder är följande.
jag. Spektroskopiska metoder(utan fotografi) nästan alla är fotometriska metoder. Dessa inkluderar: 1) Barratt-metoden. Samtidigt exciteras spektra av två ämnen - testet och standarden - synliga i spektroskopets synfält sida vid sida, den ena ovanför den andra. Strålarnas väg visas i fig. 14,

där F 1 och F 2 är två gnistgap, varifrån ljuset passerar genom Nicolas prismor N 1 och N 2 och polariserar strålarna i ömsesidigt vinkelräta plan. Med hjälp av prisma D kommer strålarna in i spektroskopets slits S. Ett tredje Nicolas-prisma, en analysator, placeras i dess teleskop, roterande vilket uppnår samma intensitet av de två linjerna som jämförs. Tidigare, när man studerar standarder, det vill säga ämnen med känt innehåll av element, fastställs förhållandet mellan analysatorns rotationsvinkel och koncentrationen, och ett diagram ritas från dessa data. Vid analys med analysatorns rotationsvinkel hittas den önskade procentandelen från detta diagram. Metodens noggrannhet är ±10%. 2) . Principen för metoden är att ljusstrålar efter spektroskopprismat passerar genom ett Wollaston-prisma, där de divergerar i två strålar och polariseras i inbördes vinkelräta plan. Strålvägsdiagrammet visas i fig. 15,

där S är slitsen, P är spektroskopprismat, W är Wollastonprismat. I synfältet erhålls två spektra B 1 och B 2, som ligger bredvid varandra; L - förstoringsglas, N - analysator. Om du roterar Wollaston-prismat kommer spektra att röra sig i förhållande till varandra, vilket gör att du kan kombinera två av deras linjer. Till exempel, om järn innehållande vanadin analyseras, kombineras vanadinlinjen med någon närliggande enfärgad järnlinje; sedan, genom att vrida på analysatorn, uppnår de samma ljusstyrka på dessa linjer. Analysatorns rotationsvinkel, som i föregående metod, är ett mått på koncentrationen av det önskade elementet. Metoden är särskilt lämplig för analys av järn, vars spektrum har många linjer, vilket gör det möjligt att alltid hitta linjer lämpliga för forskning. Metodens noggrannhet är ± (3-7)%. 3) Occhialini-metoden. Om elektroderna (till exempel metallerna som analyseras) placeras horisontellt och bilden projiceras från en ljuskälla på den vertikala slitsen i spektroskopet, kan det uppstå linjer av föroreningar både under gnist- och bågarladdningar. öppna beroende på koncentrationen på ett större eller mindre avstånd från elektroderna. Ljuskällan projiceras på slitsen med hjälp av en speciell lins utrustad med en mikrometrisk skruv. Under analysen rör sig denna lins och bilden av ljuskällan rör sig tillsammans med den tills eventuell föroreningslinje i spektrumet försvinner. Måttet på föroreningskoncentration är avläsningen på linsskalan. För närvarande har denna metod även utvecklats för att arbeta med den ultravioletta delen av spektrumet. Det bör noteras att Lockyer använde samma metod för att belysa slitsen i en spektralapparat och han utvecklade en metod för kvantitativ spektralanalys, den så kallade. "långa och korta linjer"-metoden. 4) Direkt fotometri av spektra. Metoderna som beskrivs ovan kallas visuella. Istället för visuella studier använde Lundegaard en fotocell för att mäta intensiteten hos spektrallinjer. Noggrannheten för att bestämma alkalimetaller när man arbetar med en låga nådde ± 5%. För gnisturladdningar är denna metod inte tillämplig, eftersom de är mindre konstanta än lågor. Det finns också metoder som bygger på att ändra induktansen i sekundärkretsen, samt att använda artificiell dämpning av ljuset som kommer in i spektroskopet tills de spektrallinjer som studeras försvinner i synfältet.
II. Spektrografiska metoder. Med dessa metoder undersöks fotografiska fotografier av spektra, och måttet på intensiteten hos spektrallinjerna är den svärtning de producerar på den fotografiska plattan. Intensiteten bedöms antingen med ögat eller fotometriskt.
A. Metoder utan fotometri. 1) Sista radernas metod. När koncentrationen av något element i spektrumet ändras, ändras antalet av dess linjer, vilket gör det möjligt, under konstanta driftsförhållanden, att bedöma koncentrationen av det element som bestäms. En serie spektra av ämnen med känt innehåll av den intressanta komponenten fotograferas, antalet linjer bestäms på spektrogrammen och tabeller sammanställs som anger vilka linjer som är synliga vid givna koncentrationer. Dessa tabeller tjänar vidare för analytiska definitioner. Vid analys av spektrogrammet bestäms antalet rader av det intressanta elementet och procenthalten hittas från tabellerna, och metoden ger inte en entydig siffra, utan koncentrationsgränser, det vill säga "från-till". Det är mest tillförlitligt möjligt att särskilja koncentrationer som skiljer sig från varandra med en faktor 10, till exempel från 0,001 till 0,01 %, från 0,01 till 0,1 %, etc. Analytiska tabeller är viktiga endast för mycket specifika driftsförhållanden, som kan variera mycket mellan laboratorier; Dessutom krävs noggrann efterlevnad av konstanta arbetsförhållanden. 2) Jämförande spektrametod. Flera spektra av det analyserade ämnet A + x% B fotograferas, där innehållet av x element B bestäms, och i intervallen mellan dem på samma fotografiska platta - spektra av standardämnen A + a% B, A + b % B, A + c% B , där a, b, c är den kända procentandelen av B. I spektrogrammen bestämmer intensiteten på B-linjerna mellan vilka koncentrationer värdet av x ligger. Kriteriet för driftsförhållandenas konstantitet är intensitetslikheten i alla spektrogram för någon närliggande linje A. När man analyserar lösningar läggs samma mängd av något element till dem, vilket ger en linje nära linjer B, och sedan konstansen av driftsförhållandena bedöms av lika intensitet i dessa linjer. Ju mindre skillnaden är mellan koncentrationerna av a, b, c, ... och ju mer exakt intensitetslikheten för linje A uppnås, desto mer exakt blir analysen. A. Ris använde till exempel koncentrationer av a, b, c, ..., relaterade till varandra, som 1: 1,5. Intill metoden för jämförande spektra finns metoden för "selektion av koncentrationer" (Testverfahren) enligt Güttig och Thurnwald, som endast är tillämplig på analys av lösningar. Det ligger i det faktum att om i två lösningar som innehåller a% A och x% A (x är större eller mindre än a), som nu kan bestämmas från deras spektra, så läggs en sådan mängd n av grundämnet A till varje av dessa lösningar så att intensiteten på dess linjer i båda spektra blir densamma. Detta kommer att bestämma koncentrationen x, som kommer att vara lika med (a ± n)%. Du kan också lägga till något annat element B till den analyserade lösningen tills intensiteterna för vissa linjer A och B är lika och, baserat på mängden B, uppskatta innehållet av A. 3) Homolog parmetod. I spektrumet av ett ämne A + a% B är linjerna för elementen A och B inte lika intensiva och, om det finns ett tillräckligt antal av dessa linjer, kan du hitta två sådana linjer A och B, vars intensitet kommer att vara samma. För en annan sammansättning A + b% B kommer andra linjer A och B att ha lika intensitet, etc. Dessa två identiska linjer kallas homologa par. Koncentrationerna av B där ett eller annat homologt par uppstår kallas fästpunkter detta par. För att arbeta med denna metod krävs en preliminär sammanställning av tabeller över homologa par med användning av substanser med känd sammansättning. Ju mer kompletta tabellerna är, det vill säga ju fler homologa par de innehåller med fästpunkter som skiljer sig så lite som möjligt från varandra, desto mer exakt blir analysen. Ett ganska stort antal av dessa tabeller har sammanställts, och de kan användas i vilket laboratorium som helst, eftersom förhållandena för utsläppen under deras sammanställning är exakt kända och dessa förhållanden kan användas. helt korrekt återgiven. Detta uppnås med följande enkla teknik. I spektrumet av ämne A + a% B väljs två linjer av element A, vars intensitet varierar mycket beroende på värdet av självinduktion i sekundärkretsen, nämligen en båglinje (tillhör den neutrala atomen) och en gnistlinje (tillhör jonen). Dessa två linjer kallas fixerande par. Genom att välja värdet på självinduktans görs linjerna i detta par identiska och kompileringen utförs exakt under dessa förhållanden, alltid indikerade i tabellerna. Under samma förhållanden utförs analysen och procentandelen bestäms baserat på implementeringen av ett eller annat homologt par. Det finns flera modifikationer av metoden med homologa par. Av dessa är den viktigaste metoden hjälpspektrum, används när elementen A och B inte har tillräckligt många rader. I det här fallet är spektrallinjerna för element A på ett visst sätt förbundna med linjerna för ett annat, mer lämpligt element G, och rollen som A börjar spelas av element G. Metoden för homologiska par utvecklades av Gerlyach och Schweitzer. Den är tillämpbar på både legeringar och lösningar. Dess noggrannhet är i genomsnitt cirka ±10 %.
I. Metoder som använder fotometri. 1) Barratt-metoden. Fikon. 16 ger en uppfattning om metoden.

F 1 och F 2 är två gnistgap, med hjälp av vilka spektra för standarden och det analyserade ämnet exciteras samtidigt. Ljus passerar genom 2 roterande sektorer S 1 och S 2 och bildar med hjälp av ett prisma D spektra som ligger ovanför varandra. Genom att välja sektorskärningar ges linjerna i det element som studeras samma intensitet; koncentrationen av det element som bestäms beräknas från förhållandet mellan värdena på sticklingarna. 2) är liknande, men med ett gnistgap (Fig. 17).

Ljus från F delas upp i två strålar och passerar genom sektorerna S 1 och S 2, med hjälp av Hüfner-rhombus R, två remsor av spektrumet erhålls ovanför varandra; Sp - spektrografslits. Sektorsnitten ändras tills intensiteten av föroreningslinjen och eventuell närliggande linje av huvudämnet är lika, och den procentuella halten av det element som bestäms beräknas från förhållandet mellan skärvärdena. 3) När den används som fotometer roterande logaritmisk sektor linjerna får ett kilformat utseende på spektrogrammen. En av dessa sektorer och dess position i förhållande till spektrografen under drift visas i fig. 18, a och b.

Sektorskärningen följer ekvationen
- log Ɵ = 0,3 + 0,2l
där Ɵ är längden på bågen i delar av en hel cirkel, belägen på ett avstånd I, mätt i mm längs radien från dess ände. Ett mått på intensiteten hos linjerna är deras längd, eftersom med en förändring i koncentrationen av ett element ändras också längden på dess kilformade linjer. Först, med hjälp av prover med känt innehåll, konstrueras ett diagram av beroendet av längden på en linje på %-halten; När den analyseras på ett spektrogram mäts längden på samma linje och procentsatsen hittas från diagrammet. Det finns flera olika modifieringar av denna metod. Det är värt att påpeka modifieringen av Scheibe, som använde den så kallade. dubbel logaritmisk sektor. En vy av denna sektor visas i fig. 19.

Linjerna undersöks sedan med en speciell apparat. Noggrannhet uppnåbar med logaritmiska sektorer, ±(10-15)%; Scheibes modifiering ger en noggrannhet på ±(5-7)%. 4) Ganska ofta används fotometri av spektrallinjer med ljus och termoelektriska spektrofotometrar av olika design. Termoelektriska fotometrar, designade speciellt för kvantitativ analys, är bekväma. Till exempel i FIG. Figur 20 visar ett diagram över fotometern enligt Sheibe:

L är en konstant ljuskälla med en kondensor K, M är en fotografisk platta med det spektrum som studeras, Sp är en slits, O 1 och O 2 är linser, V är en slutare, Th är ett termoelement som är kopplat till galvanometern . Ett mått på intensiteten hos linjerna är avböjningen av galvanometernålen. Mindre vanligt förekommande är självregistrerande galvanometrar, som registrerar intensiteten på linjer i form av en kurva. Analysnoggrannheten vid användning av denna typ av fotometri är ±(5-10)%. I kombination med andra metoder för kvantitativ analys kan noggrannheten vara ökat; till exempel, tre radsmetod Scheibe och Schnettler, som är en kombination av den homologa parmetoden och fotometriska mätningar, kan i gynnsamma fall ge en noggrannhet på ±(1-2)%.
Spektralanalysmetoder är baserade på studiet av optiska emissions- eller absorptionsspektra. Man skiljer på atomabsorptionsspektralanalys (analys baserad på absorptionsspektra) och emissionsspektralanalys (analys baserad på emissionsspektra). Spektralanalys används i stor utsträckning för kvalitativ och kvantitativ analys av olika ämnen. Från de karakteristiska linjerna i spektrumet kan grundämnessammansättningen av ett ämne bestämmas, och intensiteten på spektrallinjen är ett mått på koncentrationen av ämnet i provet.
Emissionsspektroskopi
Atomer av element i ett exciterat tillstånd avger strålning med en strikt definierad våglängd. Emissionsspektra (emissionsspektra) för varje grundämne är individuella, de består av en viss uppsättning karakteristiska linjer, från vilka ämnets grundämnessammansättning och dess koncentration kan bestämmas.
Vid emissionsspektralanalys förångas eller bränns provet som studeras om det är en vätska eller fast substans, och utsätts sedan för hög temperatur eller en elektrisk laddning för att överföra atomerna till ett exciterat tillstånd, och spektrumet registreras. Kvalitativ emissionsanalys handlar om att dechiffrera linjerna i det analyserade provets spektrum. Kvantitativ analys är baserad på att jämföra intensiteten hos provets spektrallinjer med intensiteten hos linjerna i ett standardprovs spektrum, varvid innehållet av elementet bestäms i vilket är känt.
Excitationskällor kan vara låga, ljusbåge, gnista, puls eller elektrisk vakuumurladdning. En ljusbågsurladdning ger en temperatur på 5000-7000 °C, vid vilken atomer av de flesta grundämnen går in i ett exciterat tillstånd. I en högspänningsgnista med en temperatur på 7000-15000 °C exciteras atomer av element med hög excitationspotential. Puls- och elektriska vakuumurladdningar används för att excitera inerta gaser.
Enligt metoden för spektrumregistrering särskiljs flera typer av emissionsspektralanalys. Vid visuell analys bestäms den kvalitativa sammansättningen genom direkt observation av det synliga spektrumet. Mer exakt är fotografisk analys, enligt vilken spektrumet fotograferas på en fotografisk platta, som sedan undersöks på en spektroprojektor för kvalitativa bestämningar eller fotometeras med hjälp av en mikrofotometer för kvantitativa bestämningar. En fast serie av linjer som motsvarar spektrallinjerna i provet som studeras erhålls på en fotografisk platta, vars svärtningsgrad är proportionell mot intensiteten av dessa linjer.
Spektroprojektorer används för att dechiffrera spektrogram. Den inhemska industrin producerar PS-18-spektroprojektorn, som gör det möjligt att få små delar av spektrumet förstorade 20 gånger på skärmen, vilket gör det lättare att dechiffrera dem under uttrycklig kvalitativ eller semikvantitativ analys.
Densiteten för svärtning av linjer på en fotografisk platta mäts med hjälp av mikrofotometrar. Ljusflödet leds genom den oswartade delen av den fotografiska plattan och riktas sedan till en fotocell med en galvanometer. Avböjningen av galvanometernålen på skalan noteras. Därefter leds ljusflödet genom den svärtade delen av plattan och avböjningen av galvanometernålen noteras återigen. Svartningsdensiteten bestäms av ekvationen:
där I0 är intensiteten av ljus som passerar genom den oförsvarta delen av den fotografiska plattan; I är intensiteten av ljus som passerar genom den svärtade delen av den fotografiska plattan.
Eftersom svärtningens täthet är proportionell mot koncentrationen av elementet, konstrueras en kalibreringsgraf över svärtningens beroende av koncentrationen baserat på galvanometerns avläsningar. Med hjälp av denna graf bestäms sedan innehållet i elementet. För att bestämma tätheten för svärtning av linjer på ett spektrogram används en MF-2 (eller MF-4) mikrofotometer och en IFO-451 tvåstrålsmikrofotometer.
Vid fotoelektrisk emissionsanalys registreras analytiska linjer med hjälp av fotoceller. Resultatet av analysen anges på mätanordningens skala eller spelas in på bandet av en självregistrerande anordning.
Kvartsspektrograf ISP-28. ISP-28-spektrografen används för att erhålla spektra i våglängdsområdet 200-600 nm. Den genomför kvalitativa och kvantitativa analyser av metaller, legeringar, malmer, mineraler och andra material. I fig. 126 visar det optiska diagrammet för anordningen. Ljus från källa 1 (båge eller gnista) genom en kondensor 3-5 med tre linser, skyddad från metallstänk av en kvartsplatta 2, riktas in i en slits 6 placerad i fokus för en spegellins 8. En parallell ljusstråle reflekterat från denna lins riktas mot ett kvartsprisma 9. Det exponerade dispersionsljuset fokuseras av en kvartslins 10 på emulsionen av den fotografiska plattan 11.

Andra spektrografer. Bänken kvarts laboratoriespektrograf ISP-30 används för kvalitativ analys av metaller, legeringar och malmer; Glasspektrografen med tre prisma ISP-51 används för analys av ämnen som innehåller grundämnen med ett litet antal spektrallinjer. För att analysera ämnen som innehåller grundämnen med särskilt komplexa spektra används STE-1-spektrografen. För kvalitativ och kvantitativ analys av metaller, malmer, mineraler etc. används en långfokusspektrograf DFS-8 (tre modifieringar) med diffraktionsgitter och en diffraktionsspektrograf DFS-452.
Flamfotometri
Flamfotometri är en av de mest exakta metoderna för emissionsspektralanalys. Denna metod används i stor utsträckning för bestämning av alkali- och jordalkalimetaller. Kärnan i flamfotometrimetoden är som följer.
Lösningen av det analyserade ämnet sprutas med tryckluft in i flamzonen på en gasbrännare, i vilken acetylen, väte, belysning eller någon annan gas förbränns. Brännarlågan fungerar också som en energikälla för att excitera atomer. En optisk anordning väljer spektrallinjen för det element som bestäms och mäter dess intensitet med hjälp av en fotocell. Spektrallinjens intensitet är proportionell mot saltkoncentrationen i lösningen (inom vissa gränser). Koncentrationen av grundämnet bestäms med hjälp av en kalibreringskurva. Nedan är sammansättningen av vissa brandfarliga gasblandningar och medeltemperaturen som erhålls vid förbränning (i °C):

Bärbar flamfotometer PPF-UNIZ. Det schematiska diagrammet för PPF-UNIZ-fotometern visas i fig. 127. Brännbar gas från en cylinder (eller stadsnät) passerar genom manostat 2, buffertflaska 3, filter 4 och kommer in genom mikrokranen 5 in i blandaren 7, som samtidigt utför funktionen som en droppavskiljare. Gastrycket efter manostaten hålls konstant med hjälp av en mikrokran 5 och mäts med en U-formad vätsketrycksmätare 6. Överskottsgas kommer ut i laboratoriebrännaren 1 och förbränns.

Komprimerad luft från en kompressor (utan användning av oljesmörjning) eller från en cylinder kommer in i en 3" buffertflaska och sedan in i ett filter 13. Lufttrycket hålls konstant med hjälp av en mikrokran 12 och mäts med en tryckmätare 11. Luften kommer in i sprutan 8, där den analyserade lösningen sugs från glaset 10. Lösningen i form av en finfördelad aerosol kommer in i blandare 7, där den blandas med brandfarlig gas.. Gas-luftblandningen som lämnar blandaren, innehållande elementet under studie i sprutat tillstånd, går in i brännaren 20 genom en droppavskiljare 14.
Våglängden för den gula flamlinjen av natrium är 589±5 µm, den röda linjen av kalcium är 615±5 µm och den infraröda linjen av kalium är 766±5 µm. Intensiteten av dessa linjer registreras av en fotocell 16, utrustad med utbytbara interferensfilter 17 och membran 18. Vid bestämning av natrium och kalcium används selenfotoceller av typen AFI-5 med en känslighet på 460-500 μA/lm, t.ex. bestämningen av kalium - en silversvavelfotocell av typen FESS-UZ med känslighet 6000-9000 µA/lm. Fotoceller och ljusfilter skyddas från direkt värmestrålning från lågan av en glasskärm 19. De resulterande fotoströmmarna registreras av en magnetoelektrisk mikroamperemeter 21 typ M-95, till vilken två av de tre fotocellerna är anslutna enligt en kompensationskrets genom en elektrisk strömbrytare 15.
Innan du börjar arbeta med enheten, öppna luckan 10 (bild 128) och säkra den med en spärr. Ett gummirör ansluts till sprutans 12 dräneringsrör 14 och sänks ned i ett kärl med en spärrvätska 20-25 cm hög. Ett glas med en kapacitet på 25-30 ml destillerat vatten placeras under sugröret 13 av sprutan. En skyddsanordning (visir) 11 är installerad på dörren och anordningen är ansluten till ett växelströmsnät på 220 V (50 Hz). Slå på kompressorn för att tillföra luft och, genom att långsamt vrida mikrokranens handtag "luft" 4 moturs, uppnå en bra finfördelning av destillerat vatten, d.v.s. bildning av starkt dispergerad aerosol. Det optimala lufttrycket (4-8) * 10000 Pa (0,4-0,8 atm) bör inte ändras under hela mättiden.

Vrid långsamt handtaget på mikrokranen "gas" 5, tillför gas till brännaren och efter 10-20 s, tänd den vid ingången till brännaren och vid utloppet av manostaten. Gastillförseln justeras så att lågans inre kon är grönmålad och den yttre är blåblå. Använd handtag 9 för att ställa in brännaren i ett läge där lågans inre kon sänks 5-6 cm under kanten av membraninloppet.
Mätningarna börjar efter 20 minuters uppvärmning av den fotometriska cellen. Under uppvärmningsperioden måste cellmembranet vara helt öppet, mikroamperemetern sätts på till låg känslighet (1,0 μA) och destillerat vatten införs i brännarlågan. Efter att ha värmt upp den fotoelektriska cellen stängs membranet, handtaget på mikroamperemetern 6 växlas till högsta känslighet (0,1 μA) och mikroamperemeterpekaren ställs in på noll genom att vrida korrigeringshuvudet på enhetens högra sida.
För att konstruera en kalibreringskurva förbereds en serie standardlösningar. För att bereda stamlösningen löses 2,385 g kaliumklorid KCl (reagenskvalitet) i en 500 ml mätkolv och späds med vatten till märket. Pipettera 5,00 ml av denna lösning till en 500 ml mätkolv och späd med destillerat vatten till märket (100-faldig utspädning). Den resulterande lösningen innehåller 25 mg kalium i 1 ml; lösningar som innehåller 5, 10, 15 och 20 mg kalium i 1 ml framställs av den. För att göra detta, pipettera 20, 40, 60 och 80 ml av en lösning innehållande 25 mg/ml kalium i 100 ml mätkolvar och späd volymen med vatten till märket.
Dessa lösningar införs sekventiellt i brännarlågan och mikroamperemeteravläsningarna registreras. När du flyttar från en lösning till en annan tvättas sprutan med destillerat vatten tills mikroampernålen återgår till noll. Baserat på erhållna data, konstrueras en kalibreringsgraf: mikroamperemeteravläsningar (längs abskissaxeln) - koncentration av det element som bestäms (längs ordinataaxeln) (i mg/ml).
För att bestämma koncentrationen av ett grundämne i testlösningen förs det in i brännarlågan och mikroamperemeteravläsningarna registreras, från vilka man, med hjälp av en kalibreringsgraf, bestämmer koncentrationen av det element som bestäms. Under hela analysprocessen är det nödvändigt att upprätthålla konstant luft- och gastryck.
Förutom metoden att bestämma koncentration med hjälp av en kalibreringskurva används metoden för att begränsa lösningar, d.v.s. ta avläsningar av en mikroamperemeter när du analyserar lösningen som studeras och, parallellt, avläsningar från enheten vid analys av standardlösningar: lösningar med lägre och högre koncentrationer. Kaliumhalten (i mg/l) beräknas med hjälp av formeln
![]()
där cl är kaliumhalten i en mer koncentrerad standardlösning; c2 - kaliumhalt i en mindre koncentrerad standardlösning; I1 - mikroamperemeteravläsningar vid analys av en standardlösning med högre koncentration; I2 - mikroamperemeteravläsningar vid analys av en standardlösning med lägre koncentration; Ix - mikroammeteravläsningar vid analys av testlösningen.
Flamfotometer Flapho-4. Tvåkanalsapparat för seriell bestämning av natrium, kalium, kalcium, litium och bly med hög känslighet. Tillverkad i DDR.
Testlösningen av provet absorberas genom att strömma igenom; spraya med tryckluft och förvandlas till en aerosol. Aerosolen går in i en speciell tank, där en brandfarlig gas (acetylen eller propan) blandas med den, och den resulterande blandningen tillförs en brännare omgiven av renad luft. I en gaslåga avdunstar ämnet som studeras och dess atomer exciteras. Ett metalliserat interferensfilter separerar en monokromatisk strålningskomponent från det allmänna flamspektrumet, som träffar selenfotocellen. Den resulterande intermittenta fotoströmmen förstärks och matas till en mät- eller registreringsanordning. Enhetsdiagrammet visas i fig. 129.

Andra flamfotometrar: trekanalig flamfotometer FP-101 för bestämning av koncentrationen av Na, K, Ca och Li; flamfotometer PFM för kvantitativ bestämning av koncentrationer av alkaliska och alkaliska jordartsmetaller, såväl som magnesium, bor, krom och mangan; flamfotometriska vätskeanalysatorer PAZH-1 och BIAN-140 för bestämning av mikrokvantiteter av K, Na, Ca och Li i lösningar, flamfotometer för bestämning av Na och K i biologiska vätskor.
Atomabsorptionsspektrofotometri
Fria atomer i ett oexciterat tillstånd belägna i lågtemperaturflamzonen har förmågan att selektivt absorbera ljus. Våglängden för ljus som absorberas av atomerna i ett element är samma som våglängden för ljuset som sänds ut av atomerna i det elementet. Följaktligen, med hjälp av de karakteristiska linjerna i absorptionsspektrumet och deras intensitet, är det möjligt att analysera ämnen, bestämma deras sammansättning och koncentrationen av dess beståndsdelar.
För att utföra atomabsorptionsanalys avdunstas ämnet som studeras genom att det matas in i en lågtemperaturflamzon. Molekylerna i det förångade ämnet dissocierar till atomer. Ljusflödet, i vars spektrum det finns en ljuslinje som absorberas av ämnet, som passerar genom denna låga, försvagas, och ju större koncentrationen av det analyserade ämnet är, desto mer.
I fig. 130 visar ett schematiskt diagram av anläggningen för atomabsorptionsanalys. Ljus från urladdningsröret 1 (ihålig katod) passerar genom brännarens 2 låga och fokuseras på slitsen i monokromatorn 3. Därefter träffar strålningen fotomultiplikatorröret, eller fotocellen 4. Monokromatorn väljer strålning med en våglängd som absorberas av elementet som studeras från det totala ljusflödet. Strömmen förstärks i block 5 och registreras av mätanordning 6.

Bestämningen består av att mäta förhållandet mellan ljusintensiteterna som passerar genom lågan med och utan analyten införd i den. Eftersom intensiteten av spektrallinjen för elementet som studeras i brännarflamman visar sig vara större än deras strålningsintensitet från den ihåliga katoden, moduleras strålningen från den senare. Modulering av strålning (ändring av svängningarnas amplitud och frekvens) utförs med hjälp av en roterande skiva med hål (modulator 7) placerad mellan den ihåliga katoden och lågan. Förstärkaren 5 måste ha en maximal förstärkning för samma frekvens som strålningen från den ihåliga katoden moduleras med.
Atomabsorptionsspektrofotometer AAS-1. Avsedd för absorptions- och emissionsspektralanalys. Låter dig definiera 65 element.
Funktionsprincip. Det flytande provet finfördelas med en oxiderande gas, blandas med en brandfarlig gas (acetylen eller propan) och bränns i en brännarlåga. Strålning från en ihålig katodlampa passerar genom brännarlågan. Efter att ha valt en lämplig linje med en diffraktionsmonokromator riktas strålningen till en fotomultiplikator. Likströmskomponenten som orsakas av självstrålning undertrycks. Signalen från fotomultiplikatorn förstärks, likriktas av en känslig likriktare och registreras. Enheten justeras och styrs med standardlösningar.
I fig. 131 visar ett diagram över AAS-1.

Enhetsdesign. Enheten har ett beslagskomplex för tillförsel av gaser, ett sprut- och förbränningssystem, en utbytbar anordning för lampor med ihåliga katoder, ett optiskt system och en mottagningsanordning med en förstärkare och indikator.
Brännarlågan drivs av en blandning av acetylen eller propan och tryckluft. Gaser kommer in i förbränningssystemet från konventionella cylindrar med justerade (primära) tryckreducerare. Tillförseln av oljefri luft tillhandahålls av en membrankompressor (16 l/min vid ett tryck på 3*100000 Pa (3 atm)). Enhetens ventilkomplex har justerbara (sekundära) växellådor och flödesmätare för att styra flödet av varje gas, samt keramiska sintrade dammfilter och en flaska för ytterligare acetylensköljning. Säkerhetsventilen stoppar automatiskt åtkomsten av brandfarlig gas när tryckluftens driftstryck minskar (till exempel på grund av knäckning eller sönderrivning av tillförselslangen); ventilen eliminerar den felaktiga ordningen för gastillförseln vid antändning av lågan.
Finfördelnings- och förbränningssystemet är placerat bakom ett löstagbart laminerat glasfönster så att systemet kan observeras. Den ringformade munstycksförstöraren har ett högt finfördelningsförhållande och kännetecknas av lågt vätskeflöde (3,4 ml/min, eller 0,5 ml under hela analysen). Brännaren är utrustad med utbytbara munstyckshuvuden - en slitsad för absorptionsanalys (Fig. 132, a) och två flerhål (Mecker-brännare med nät) för emissionsanalys (Fig. 132,6).

Justerbara hållare för fyra ihåliga katodlampor är placerade i en enhet som möjliggör snabba lampbyten. Efter byte av en av lamporna behöver hållarna inte justeras.
Det optiska systemet riktar lampstrålningen i form av en smal stråle mot lågan. På grund av den laterala förskjutningen av röret med bildbehandlingssystemet passerar strålning genom lågan en eller tre gånger för att öka analysens känslighet. En monokromator med hög aperturdiffraktion väljer den önskade resonanslinjen från linjespektrumet för en given ihålig katodlampa. Monokromatorslitsens bredd justeras från 0 till 2 mm.
Ett precisionsdiffraktionsgitter med 1300 linjer per 1 mm och en vinkelspridning på 1,5 nm/mm har hög upplösning. Spektralområdet för gittret är från 190 till 820 nm.
Strålningsmottagaren är en 12-stegs fotomultiplikator. Instrumentförstärkaren, strömförsörjningen för den ihåliga katodlampan och fotomultiplikatorerna fungerar på transistorer och kan kompensera för nätspänningsfluktuationer från +10 till -15 %.
Enhetsavläsningarna mäts med hjälp av en mätklocka som har tre skalor: en logaritmisk skala för extinktionskoefficienten från 0 till 1,5; linjär skala från 0 till 100 och driftspänningsskala från 0 till 16 mV. En inspelnings- eller datorenhet kan vara ansluten till enheten för att bestämma koncentrationen eller för att bearbeta data. Känsligheten för bestämningar (i mg/l) är:
![]()
Enheten drivs från ett växelströmsnätverk på 220 V, 50 Hz. Tillverkad i DDR.
Andra inhemska: Atomabsorptionsspektrofotometer S-302 för bestämning av spårmängder av järn, koppar, zink, kobolt, nickel, vismut, kalcium och andra grundämnen; automatiserad atomabsorptionsspektrofotometer AA-A för bestämning av kalcium och koppar med ökad känslighet; "Saturnus" - halvautomatisk inspelningsspektrofotometer för låga atomabsorption för bestämning av 32 element; "Spectrum-1" är en atomabsorptionsspektrofotometer för snabb bestämning av mer än 40 grundämnen med en känslighet på cirka 0,2 μg/ml.
Perkin-Elmer atomabsorptionsspektrofotometer, modell 603, tillverkas i England. Enheten är byggd med ett tvåstrålsschema och kombinerat med en mikrodator. Ger hög noggrannhet och snabb bestämning. En brandfarlig syre-acetylenblandning används för att antända lågan.
Spektra, metoder för att erhålla dem, egenskaper, klassificering och användning för analytiska ändamål. Grundläggande element i spektrala instrument och deras syfte
Spektralanalysmetoder är metoder som bygger på att bestämma ämnens kemiska sammansättning och struktur utifrån deras spektrum.
Spektrum av ett ämne är den elektromagnetiska strålning som sorteras efter våglängd, emitteras, absorberas, sprids eller bryts av ämnet. Metoder baserade på att erhålla och studera emissions(emissions)spektra av elektromagnetisk strålning (energi) kallas emission, absorption (absorption) - absorption, spridning - spridningsmetoder, refraktion - refraktiv.
Spektrum av ett ämne erhålls genom att påverka det med temperatur, elektronflöde, ljusflöde (elektromagnetisk energi) med en viss våglängd (strålningsfrekvens) och andra metoder. Vid en viss mängd stötenergi kan ett ämne gå in i ett exciterat tillstånd. I detta fall uppstår processer som leder till uppkomsten av strålning med en viss våglängd i spektrumet (tabell 2.2.1).
Emission, absorption, spridning eller brytning av elektromagnetisk strålning kan betraktas som en analytisk signal som bär information om den kvalitativa och kvantitativa sammansättningen av ett ämne eller dess struktur. Frekvensen (våglängden) av strålningen bestäms av sammansättningen av ämnet som studeras, och intensiteten av strålningen är proportionell mot antalet partiklar som orsakade dess uppkomst, d.v.s. kvantitet av ett ämne eller en komponent i en blandning.
Var och en av de analytiska metoderna använder vanligtvis inte hela spektrumet av ett ämne, som täcker våglängdsområdet från röntgenstrålar till radiovågor, utan bara en viss del av det. Spektralmetoder kännetecknas vanligtvis av spektrala våglängder som fungerar för en given metod: ultraviolett (UV), röntgen, infraröd (IR), mikrovågsugn, etc.
Metoder som fungerar inom UV, synligt och IR-området kallas optiska. De används mest i spektrala metoder på grund av den jämförande enkelheten hos utrustningen för att erhålla och registrera spektrumet.
Spektra i det optiska området är resultatet av förändringar i energin hos atomer eller molekyler.
Tabell 2.2.1
| Typ av strålning | Atom- och molekylära processer | Källor till excitation | Strålningsdetektorer | |
| , nm | namn | |||
| 10-3 | -strålning | Kärn | Cyklotroner | Geigerräknare, |
| 10-2 | Röntgen | reaktioner | scintillationsräknare, fotografiska plattor | |
| 10-1 | Övergångar av yttre | Röntgenrör | ||
| 100 | elektroner | |||
| 101 | UV vakuum | |||
| 2·102 | UV långt | Yttre elektronövergångar | Röntgenrör, gnista, låga, båge | Fotoceller, fotomaterial |
| 3·102 | UV nära | |||
| 375-750 | Synlig | Öga, fotocell | ||
| 104 | IR nära | Vibrationer av molekyler | Uppvärmda metalltrådar | Vakuum termoelement, |
| 105 | Ytterligare | Rotation av molekyler | bolometrar | |
Som ett resultat av en förändring i energin hos en atom eller molekyl, flyttar de sig från grundtillståndet med minsta möjliga inre energi E0 till ett exciterat tillstånd med energi E1. Intern energi är en diskret (kvantum) kvantitet, därför åtföljs övergången av en atom eller molekyl från grundtillståndet till en annan alltid av en abrupt förändring i energi, d.v.s. ta emot eller ge ut en del (kvantum) energi.
Kvanta av elektromagnetisk strålning är fotoner, vars energi är relaterad till strålningens frekvens och våglängd genom ett känt förhållande
E = h · =
,där E = E1 - E2, E1 är energin för initialen och E2 är energin för sluttillståndet för atomen eller molekylen mellan vilken övergången sker; h - Plancks konstant; c är ljusets hastighet; - frekvens; - våglängd för elektromagnetisk strålning.
När en atom är exciterad, rör sig elektroner från de yttre fyllda nivåerna till ofyllda högre energinivåer.
En atom kan inte förbli i ett exciterat tillstånd länge. Han strävar efter att ge bort den mottagna överskottsenergin och återgå till ett oupphetsat tillstånd. Efter en mycket kort tid (10-8 - 10-7 s) återgår atomen spontant från det exciterade tillståndet till marken eller mellantillståndet.
När en elektron passerar från en övre nivå till en lägre, frigörs en foton - ett strålningskvantum med vissa och .
Schematiskt kan elektroniska övergångar i atomer mellan olika tillstånd, åtföljda av emission och absorption av kvanta av elektromagnetisk strålning, representeras i form av ett diagram (Fig. 2.2.1).

Horisontella linjer i Fig. 2.2.1. Energinivåerna för olika tillstånd av atomen är avbildade. Nivå E0 är nivån för grundtillståndet; E1, E2, E3 - nivåer av exciterade tillstånd i ordning efter ökande energi. Vertikala pilar motsvarar emissionen (nedåtpilen) eller absorptionen () av en foton. Det är uppenbart
01 = 10, 13 = 31, etc.
Den uppsättning fotoner som emitteras eller absorberas under en elektronisk övergång av en atom, som skapar strålning med en våglängd, kallas en spektrallinje. Spektrallinjens våglängd kan bestämmas från relationen =
. Uppsättningen av spektrallinjer relaterade till en specifik atom (molekyl) bildar spektrumet för den atomen (molekylen).Spektrum som orsakas av övergången vid E1 E2 kallas emissionsspektrum, och vid E1 E2 - absorptionsspektrum. Övergångar och motsvarande spektrallinjer som passerar från eller till huvudenerginivån kallas resonans.
För att excitera en spektrallinje krävs en viss energi, kallad excitationspotential. Om man tillför för mycket energi till en atom kan ett fullständigt avlägsnande av en elektron ske, d.v.s. jonisering av atomen. Den energi som krävs för detta kallas joniseringspotential. Resonanslinjer är de ljusaste och kännetecknas av den lägsta excitationspotentialen.
En förändring i en molekyls energi åtföljs av en förändring i både vibrations- och rotationsenergin, d.v.s. molekylen har inte rent elektroniska övergångar, utan endast elektroniska-vibrations-rotationsövergångar (EV) är möjliga. Antalet möjliga ECV-övergångar i en molekyl är mycket större än i atomer, därför är molekylspektra som regel mer komplexa och består av ett större antal spektrallinjer i det optiska våglängdsområdet. Ett schematiskt diagram över energinivåerna för en molekyl kan presenteras enligt följande (Fig. 2.2.2).

Fig.2.2.2. Diagram över molekylära energinivåer
För både molekyler och atomer sker inte alla tänkbara övergångar. Övergångar regleras av så kallade urvalsregler: övergångar där kvanttalet ändras med ett är tillåtna (till exempel Sp, pd, etc.).
För analytiska ändamål kan både emissions- och absorptionsspektra användas eftersom de är relaterade till varandra. Till exempel producerar ljus som emitteras av glödande natriummetallånga som passerar genom ett prisma två mycket nära gula linjer med våglängder på 589,0 och 589,6 mikron. Dessa är de så kallade D - natriumlinjerna. Å andra sidan, om du passerar polykromatiskt vitt ljus (dvs. en uppsättning ljusstrålar med alla våglängder) genom natriumånga och sedan sönderdelar det till dess komponentfärger i ett glasprisma, då mot bakgrunden av ett kontinuerligt spektrum två svarta linjer kommer att detekteras exakt på plats D - linjer. Följaktligen absorberar natriumånga strålning exakt vid de våglängder som de avger när de exciteras.
Detta är ett allmänt mönster, så spektralanalys kan utföras med både emissionsspektrum och absorptionsspektrum. Den första metoden är bekväm för att analysera material där emissionsspektrumet av ingående ämnen, såsom metaller och gaser, lätt exciteras, och den andra metoden är bekvämare för att analysera material där det är svårt att excitera ingående ämnen (till exempel, lösningar).
Emissionsspektra är uppdelade i kontinuerlig, randig och linje (Fig. 2.2.3). Kontinuerliga (eller kontinuerliga) spektra innehåller alla våglängder i ett visst intervall.
De sänds ut av heta föremål som är belägna på sådant avstånd från varandra att deras strålning kan anses vara oberoende. Gaser och metallångor har linjespektra.
Linjerna i spektra av atomer är inte slumpmässigt placerade, utan kombineras till grupper som kallas serier. Avstånden mellan linjerna i serien minskar naturligtvis när vi går från längre till kortare vågor.
Balmeer, för det enklaste linjespektrumet av väte, upptäckte att frekvenserna för spektrallinjer i serie belägna i olika regioner av elektromagnetisk strålning står i ett visst regelbundet förhållande till varandra, vilket i allmänna termer för alla element uttrycktes av beroendet

eller i vissa fall
Spektralanalys är uppdelad i flera oberoende metoder. Bland dem är: infraröd och ultraviolett spektroskopi, atomabsorption, luminescens- och fluorescensanalys, reflektion och Raman-spektroskopi, spektrofotometri, röntgenspektroskopi, samt ett antal andra metoder.
Absorptionsspektralanalys baseras på studiet av absorptionsspektra för elektromagnetisk strålning. Emissionsspektralanalys utförs med användning av emissionsspektra av atomer, molekyler eller joner exciterade på olika sätt.
Atomemissionsspektralanalys
Spektralanalys kallas ofta enbart atomemissionsspektralanalys, som bygger på studiet av emissionsspektra av fria atomer och joner i gasfasen. Den utförs i våglängdsområdet 150-800 nm. Ett prov av ämnet som studeras införs i strålningskällan, varefter avdunstning och dissociation av molekyler sker i den, såväl som excitation av de resulterande jonerna. De avger strålning, som registreras av spektralinstrumentets inspelningsenhet.
Jobbar med Spectra
Provernas spektra jämförs med spektra för kända element, som kan hittas i motsvarande tabeller över spektrallinjer. Så bestäms sammansättningen av det ämne som analyseras. Kvantitativ analys involverar koncentrationen av ett givet element i analyten. Det känns igen av signalens storlek, till exempel genom graden av svärtning eller optisk densitet hos linjer på en fotografisk platta, eller av intensiteten av ljusflödet på en fotoelektrisk mottagare.
Typer av spektra
Ett kontinuerligt spektrum av strålning tillhandahålls av ämnen i fast eller flytande tillstånd, såväl som täta gaser. Det finns inga avbrott i ett sådant spektrum, vågor av alla längder är representerade i det. Dess karaktär beror inte bara på egenskaperna hos enskilda atomer, utan också på deras interaktion med varandra.
Ett linjeemissionsspektrum är karakteristiskt för ämnen i gasform, medan atomerna nästan inte interagerar med varandra. Faktum är att isolerade atomer av ett kemiskt element avger vågor med en strikt definierad våglängd.
När gasdensiteten ökar börjar spektrallinjerna att vidgas. För att observera ett sådant spektrum används glöden från en gasurladdning i ett rör eller ånga av ett ämne i en låga. Om vitt ljus leds genom en icke-emitterande gas, kommer mörka linjer i absorptionsspektrumet att uppträda mot bakgrund av källans kontinuerliga spektrum. Gas absorberar mest intensivt ljuset av de våglängder som den avger när den värms upp.
En av de viktigaste metoderna för att analysera den kemiska sammansättningen av ett ämne är spektralanalys. En analys av dess sammansättning utförs baserat på studien av dess spektrum. Spektralanalys - används i olika studier. Med dess hjälp upptäcktes ett komplex av kemiska element: He, Ga, Cs. i solens atmosfär. Förutom Rb, In och XI bestäms sammansättningen av solen och de flesta andra himlakroppar.
Ansökningar
Spektral expertis, vanlig inom:
- Metallurgi;
- Geologi;
- Kemi;
- Mineralogi;
- Astrofysik;
- Biologi;
- medicin osv.
Låter dig hitta de minsta mängderna av ett etablerat ämne i de föremål som studeras (upp till 10 - MS) Spektralanalys är uppdelad i kvalitativ och kvantitativ.
Metoder
Metoden för att fastställa den kemiska sammansättningen av ett ämne baserat på spektrumet är grunden för spektralanalys. Linjespektra har en unik personlighet, precis som mänskliga fingeravtryck eller mönstret av snöflingor. Det unika med mönster på huden på ett finger är en stor fördel för att söka efter en brottsling. Därför, tack vare särdragen i varje spektrum, är det möjligt att fastställa det kemiska innehållet i kroppen genom att analysera ämnets kemiska sammansättning. Även om dess massa av ett element inte överstiger 10 - 10 g, kan det med hjälp av spektralanalys detekteras i sammansättningen av ett komplext ämne. Detta är en ganska känslig metod.
Emissionsspektralanalys
Emissionsspektralanalys är en serie metoder för att bestämma den kemiska sammansättningen av ett ämne utifrån dess emissionsspektrum. Grunden för metoden att fastställa den kemiska sammansättningen av ett ämne - spektral undersökning - bygger på mönstren i emissionsspektra och absorptionsspektra. Denna metod låter dig identifiera miljondelar av ett milligram av ett ämne.
Det finns metoder för kvalitativ och kvantitativ undersökning, i enlighet med fastställandet av analytisk kemi som ämne, vars syfte är att formulera metoder för att fastställa den kemiska sammansättningen av ett ämne. Metoder för att identifiera ett ämne blir oerhört viktiga inom kvalitativ organisk analys.
Baserat på linjespektrumet av ångor av något ämne är det möjligt att bestämma vilka kemiska element som ingår i dess sammansättning, eftersom alla kemiska grundämnen har sitt eget specifika utsläppsspektrum. Denna metod för att fastställa den kemiska sammansättningen av ett ämne kallas kvalitativ spektralanalys.
Röntgenspektralanalys
Det finns en annan metod för att identifiera en kemikalie som kallas röntgenspektralanalys. Röntgenspektralanalys bygger på aktiveringen av atomerna i ett ämne när det bestrålas med röntgenstrålar, en process som kallas sekundär eller fluorescerande. Aktivering är också möjlig när den bestrålas med högenergielektroner; i detta fall kallas processen direkt excitation. Som ett resultat av elektronernas rörelse i de djupare inre elektronlagren uppstår röntgenlinjer.
Wulff-Bragg-formeln låter dig ställa in våglängderna i sammansättningen av röntgenstrålning när du använder en kristall av en populär struktur med ett känt avstånd d. Detta är grunden för bestämningsmetoden. Ämnet som studeras bombarderas med höghastighetselektroner. Den placeras till exempel på anoden av ett demonterbart röntgenrör, varefter den avger karakteristiska röntgenstrålar som faller på en kristall av en känd struktur. Vinklarna mäts och motsvarande våglängder beräknas med hjälp av formeln, efter fotografering av det resulterande diffraktionsmönstret.
Tekniker
För närvarande är alla metoder för kemisk analys baserade på två tekniker. Antingen vid det fysiska testet eller vid det kemiska testet, jämför den fastställda koncentrationen med dess måttenhet:
Fysisk
Den fysikaliska tekniken bygger på metoden att korrelera en kvantitetsenhet för en komponent med en standard genom att mäta dess fysiska egenskap, vilket beror på dess innehåll i ett prov av ämnet. Funktionsförhållandet "Egenskapsmättnad – komponentinnehåll i provet" bestäms genom försök genom att kalibrera medlen för att mäta en given fysisk egenskap enligt den komponent som installeras. Från kalibreringsgrafen erhålls kvantitativa samband, konstruerade i koordinaterna: "mättnad av en fysisk egenskap - koncentration av den installerade komponenten."
Kemisk
En kemisk teknik används i metoden för att korrelera en kvantitetsenhet för en komponent med en standard. Här används lagarna för bevarande av kvantiteten eller massan av en komponent under kemiska interaktioner. Kemiska interaktioner baseras på kemiska föreningars kemiska egenskaper. I ett prov av ett ämne utförs en kemisk reaktion som uppfyller de specificerade kraven för att bestämma den önskade komponenten, och volymen eller massan som är involverad i den specifika kemiska reaktionen av komponenterna mäts. Kvantitativa samband erhålls, sedan skrivs antalet ekvivalenter av en komponent för en given kemisk reaktion eller lagen om massans bevarande ned.
Enheter
Instrument för att analysera den fysiska och kemiska sammansättningen av ett ämne är:
- Gasanalysatorer;
- Larm för högsta tillåtna och explosiva koncentrationer av ångor och gaser;
- Koncentratorer för flytande lösningar;
- Densitetsmätare;
- Saltmätare;
- Fuktmätare och andra enheter liknande ändamål och fullständighet.
Med tiden ökar utbudet av analyserade objekt och analysens hastighet och noggrannhet ökar. En av de viktigaste instrumentella metoderna för att fastställa den atomära kemiska sammansättningen av ett ämne är spektralanalys.
NOTERA:
Priset för kemisk undersökning är inklusive skatter. Transportkostnader betalas separat.
- I kontakt med 0
- Google+ 0
- OK 0
- Facebook 0






