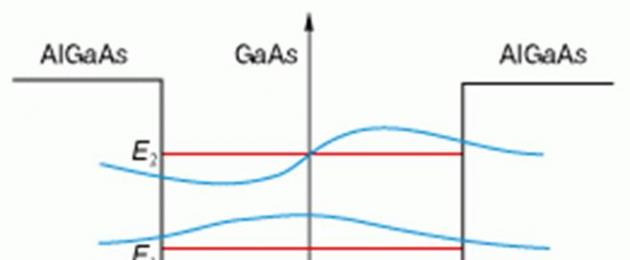
Riso. 2. Bande energetiche al confine di due semiconduttori - eterostruttura. E c E Unione Europea- confini della banda di conduzione e della banda di valenza, Per esempio- larghezza del gap di banda. Elettrone con meno energia E c 2 (livello mostrato in rosso) può trovarsi solo a destra del bordo
Per gli elettroni che si muovono in un semiconduttore a gap stretto e hanno meno energia E c 2, il confine svolgerà il ruolo di potenziale barriera. Due eterogiunzioni limitano il movimento dell'elettrone su entrambi i lati e, per così dire, formano un pozzo di potenziale.
Questo è il modo in cui vengono creati i pozzi quantistici posizionando un sottile strato di semiconduttore con una stretta banda proibita tra due strati di materiale con una banda proibita più ampia. Di conseguenza, l'elettrone viene bloccato in una direzione, il che porta alla quantizzazione dell'energia del movimento trasversale.
Allo stesso tempo, nelle altre due direzioni il movimento degli elettroni sarà libero, quindi possiamo dire che il gas di elettroni nel pozzo quantico diventa bidimensionale.
Allo stesso modo, una struttura contenente una barriera quantistica può essere preparata posizionando un sottile strato di un semiconduttore a banda larga tra due semiconduttori a banda stretta.
Sono stati sviluppati diversi processi tecnologici avanzati per la produzione di tali strutture, ma i migliori risultati nella preparazione di strutture quantistiche sono stati ottenuti utilizzando il metodo epitassia da fascio molecolare.
Per far crescere un sottile strato di semiconduttore utilizzando questo metodo, è necessario dirigere un flusso di atomi o molecole su un substrato accuratamente pulito. Diversi flussi di atomi, ottenuti facendo evaporare una sostanza da fonti riscaldate separate, volano contemporaneamente sul substrato.
Per evitare la contaminazione, la struttura viene coltivata sotto vuoto spinto. L'intero processo è controllato dal computer, composizione chimica e la struttura cristallina dello strato cresciuto vengono controllate durante il processo di crescita.
Il metodo dell'epitassia a fascio molecolare rende possibile la crescita di strati monocristallini perfetti con uno spessore di soli pochi periodi reticolari (un periodo reticolare è circa 2).
È estremamente importante che i periodi reticolari di due strati adiacenti, che hanno composizioni chimiche diverse, siano quasi gli stessi. Quindi gli strati si susseguiranno con precisione e il reticolo cristallino della struttura cresciuta non conterrà difetti.
Utilizzando il metodo dell'epitassia a fascio molecolare, è possibile ottenere un confine molto netto (preciso per un monostrato) tra due strati adiacenti e la superficie è liscia a livello atomico.
Le strutture quantistiche possono essere sviluppate da vari materiali, ma la coppia di maggior successo per la crescita dei pozzi quantistici è il semiconduttore GaAs - arseniuro di gallio e la soluzione solida Al x Ga 1-x As, in cui alcuni atomi di gallio sono sostituiti da atomi di alluminio. Grandezza Xè la frazione di atomi di gallio sostituiti da atomi di alluminio varia solitamente da 0,15 a 0,35; Il gap di banda nell'arseniuro di gallio è 1,5 eV, e nella soluzione solida Al x Ga 1-x As aumenta con l'aumentare X. Sì, quando X= 1, cioè nel composto AlAs il band gap è 2,2 eV.
Per far crescere un pozzo quantico, è necessario modificare la composizione chimica degli atomi che volano sullo strato in crescita durante la crescita.
Innanzitutto, è necessario far crescere uno strato di semiconduttore con ampio gap di banda, ovvero Al x Ga 1-x As, quindi uno strato di materiale GaAs a gap stretto e infine ancora uno strato di Al x Ga 1-x As.
Il diagramma energetico di un pozzo quantistico così preparato è mostrato in Fig. 3. Il pozzo ha una profondità finita (diversi decimi di elettronvolt). Contiene solo due livelli discreti e le funzioni d'onda al confine del pozzo non svaniscono. Ciò significa che l'elettrone può essere rilevato anche all'esterno del pozzo, nella regione dove l'energia totale è inferiore al potenziale. Naturalmente, questo non può accadere nella fisica classica, ma in fisica quantisticaè possibile.

Riso. 3. Ben formato quantistico in uno strato di semiconduttore con una stretta banda proibita inserito tra due semiconduttori con una banda proibita più ampia
I tecnologi hanno sviluppato diversi modi per produrre punti e fili quantici. Queste strutture possono essere formate, ad esempio, sull'interfaccia tra due semiconduttori dove si trova un gas di elettroni bidimensionale.
Ciò può essere fatto aggiungendo ulteriori barriere che limitano il movimento degli elettroni in una o due direzioni.
I fili quantistici sono formati sul fondo di una scanalatura a forma di V formata sul substrato semiconduttore. Se alla base di questa scanalatura viene depositato un semiconduttore con una banda proibita più piccola, gli elettroni di questo semiconduttore verranno bloccati in due direzioni.
Nella fig. La Figura 4 mostra i punti quantici creati all'interfaccia tra arseniuro di gallio e arseniuro di gallio e alluminio. Durante il processo di crescita, nel semiconduttore AlGaAs sono stati introdotti ulteriori atomi di impurità. Gli elettroni di questi atomi entrano nel semiconduttore GaAs, cioè in una regione con energia inferiore. Ma non possono andare troppo lontano, poiché sono attratti dagli atomi di impurità che hanno lasciato e che hanno ricevuto carica positiva. Quasi tutti gli elettroni sono concentrati nell'eterointerfaccia sul lato GaAs e formano un gas bidimensionale. Il processo di formazione dei punti quantici inizia depositando sulla superficie dell'AlGaAs una serie di maschere, ciascuna delle quali ha la forma di un cerchio. Successivamente viene effettuato un attacco profondo, durante il quale viene rimosso l'intero strato di AlGaAs e in parte lo strato di GaAs (in Fig. 4).

Riso. 4. Punti quantici formati in un gas di elettroni bidimensionale all'interfaccia di due semiconduttori
Di conseguenza, gli elettroni si ritrovano bloccati nei cilindri risultanti (nella Fig. 4, l'area in cui si trovano gli elettroni è colorata in rosso). I diametri dei cilindri sono dell'ordine di 500 nm.
In un punto quantico, il movimento è limitato in tre direzioni e lo spettro energetico è completamente discreto, proprio come in un atomo. Pertanto, i punti quantici sono anche chiamati atomi artificiali, sebbene ciascuno di questi punti sia costituito da migliaia o addirittura centinaia di migliaia di atomi reali.
Le dimensioni dei punti quantici (si può parlare anche di scatole quantistiche) sono dell'ordine di diversi nanometri. Come un vero atomo, un punto quantico può contenere uno o più elettroni liberi. Se c'è un elettrone, allora è come un atomo di idrogeno artificiale, se ce ne sono due, è un atomo di elio, ecc.
Punto quantico- un frammento di un conduttore o semiconduttore, limitato in tutte e tre le dimensioni spaziali e contenente elettroni di conduzione. Il punto deve essere così piccolo che gli effetti quantistici siano significativi. Ciò si ottiene se l'energia cinetica dell'elettrone ![]() , a causa dell'incertezza della sua quantità di moto, sarà notevolmente maggiore di tutte le altre scale di energia: innanzitutto maggiore della temperatura espressa in unità di energia ( D- dimensione caratteristica del punto, M- massa effettiva di un elettrone in un punto).
, a causa dell'incertezza della sua quantità di moto, sarà notevolmente maggiore di tutte le altre scale di energia: innanzitutto maggiore della temperatura espressa in unità di energia ( D- dimensione caratteristica del punto, M- massa effettiva di un elettrone in un punto).
Punto quantico Qualsiasi pezzo di metallo o semiconduttore sufficientemente piccolo può servire. Storicamente, i primi punti quantici erano probabilmente microcristalli di seleniuro di cadmio CdSe. Un elettrone in un microcristallo di questo tipo si sente come un elettrone in un pozzo di potenziale tridimensionale; ha molti livelli energetici stazionari con una distanza caratteristica tra loro; ![]() (l'esatta espressione dei livelli energetici dipende dalla forma del punto). Similmente alla transizione tra i livelli energetici di un atomo, quando un punto quantico passa da un livello energetico all'altro, può essere emesso un fotone. È anche possibile lanciare un elettrone in un livello energetico elevato e ricevere radiazione dalla transizione tra livelli inferiori (luminescenza). Inoltre, a differenza degli atomi reali, le frequenze di transizione possono essere facilmente controllate modificando le dimensioni del cristallo. In realtà, l'osservazione della luminescenza dei cristalli di seleniuro di cadmio con una frequenza di luminescenza determinata dalla dimensione del cristallo è servita come prima osservazione dei punti quantici.
(l'esatta espressione dei livelli energetici dipende dalla forma del punto). Similmente alla transizione tra i livelli energetici di un atomo, quando un punto quantico passa da un livello energetico all'altro, può essere emesso un fotone. È anche possibile lanciare un elettrone in un livello energetico elevato e ricevere radiazione dalla transizione tra livelli inferiori (luminescenza). Inoltre, a differenza degli atomi reali, le frequenze di transizione possono essere facilmente controllate modificando le dimensioni del cristallo. In realtà, l'osservazione della luminescenza dei cristalli di seleniuro di cadmio con una frequenza di luminescenza determinata dalla dimensione del cristallo è servita come prima osservazione dei punti quantici.
Attualmente, molti esperimenti sono dedicati ai punti quantici formati nel gas di elettroni bidimensionali. In un gas di elettroni bidimensionale, il movimento degli elettroni perpendicolare al piano è già limitato e una regione sul piano può essere isolata utilizzando elettrodi metallici di gate posizionati sopra l'eterostruttura. I punti quantici in un gas elettronico bidimensionale possono essere collegati tramite contatti tunnel ad altre regioni del gas bidimensionale e si può studiare la conduzione attraverso il punto quantico. In un tale sistema si osserva il fenomeno del blocco di Coulomb.

Punti quantici PbSe su strato PbTe

Riso. 1a Punto quantico di germanio a base di silicio Si 001 (foto scattata al microscopio a scansione elettronica) (disegno del gruppo di ricerca HP)

Riso. 1b Canale fotonico conico del semiconduttore come punto quantico
Gli elettroni catturati dai punti quantici si comportano come se fossero in un atomo normale, anche se l '"atomo artificiale" non ha un nucleo. Quale atomo rappresenta un tale insieme di elettroni dipende dal numero di elettroni nel punto quantico.
 Riso. Dimensioni di un punto quantico di nanocristallo
Riso. Dimensioni di un punto quantico di nanocristallo
Oltre a disegnare semplicemente un motivo sulla superficie di un semiconduttore e ad inciderlo, è possibile creare punti quantici sfruttando la proprietà naturale del materiale di formare piccole isole durante il processo di crescita. Tali isole possono, ad esempio, formarsi spontaneamente sulla superficie di uno strato cristallino in crescita. Esistono altre tecnologie per preparare pozzi quantistici, fili e punti, che a prima vista sembrano molto semplici.
Capitolo 10. CONCETTO DI TEORIA DELLE BANDE DEI SOLIDI
L'idea di valenza come capacità di un atomo di formare legami chimici con un certo numero di altri atomi quando applicata a un corpo solido perde il suo significato, poiché qui si realizza la possibilità di interazione collettiva. Quindi in una molecola la valenza degli atomi è uguale a uno, ma in un cristallo ogni atomo è circondato da 6 atomi e viceversa.
Lo spettro energetico di un atomo isolato è determinato dall'interazione degli elettroni con il nucleo ed è di natura discreta. Gli stati energetici degli elettroni in un solido sono determinati dalla sua interazione sia con il suo nucleo che con i nuclei di altri atomi. In un cristallo, i nuclei degli atomi si trovano periodicamente lungo qualsiasi direzione (Fig. 56). Pertanto, l'elettrone si muove in un campo elettrico periodico (vicino ai nuclei energia potenziale gli elettroni sono più piccoli che nello spazio tra i nuclei). Ciò porta al fatto che invece di un livello di energia atomica discreto in un solido contenente N nascono gli atomi N livelli energetici situati uno vicino all'altro, che formano una zona energetica. In questo senso si parla della suddivisione del livello energetico in una zona energetica. I livelli energetici adiacenti nella zona sono separati l'uno dall'altro di 10 -23 eV. Per confronto, segnaliamo che la media energia termica elettroni a temperatura T= 300 K è ~ 10 -2 eV. Di conseguenza, lo spettro degli elettroni all'interno della banda può essere considerato quasi continuo.
Il numero di stati in una zona è uguale al prodotto del numero di atomi nel cristallo e al multiplo del livello di energia atomica da cui si è formata la zona. Il multiplo di un livello energetico si riferisce al numero di elettroni che a questo livello possono essere soggetti al principio di Pauli.
Le zone di energie consentite sono separate da zone di energie proibite. La loro larghezza è paragonabile alla larghezza delle bande energetiche consentite. All'aumentare dell'energia, la larghezza delle bande consentite aumenta e la larghezza delle bande vietate diminuisce (Fig. 57).
§2. Metalli, semiconduttori, dielettrici
Differenze nelle proprietà elettriche solidi sono spiegati dal diverso riempimento delle bande energetiche consentite da parte degli elettroni e dall'ampiezza delle bande proibite. Affinché il corpo possa condurre corrente elettricaè necessario avere livelli di energia libera nelle bande consentite verso le quali gli elettroni potrebbero spostarsi sotto l'influenza di un campo elettrico.
Metalli
Consideriamo un cristallo di sodio. La sua formula elettronica è. Il diagramma energetico del sodio è mostrato in Fig. 58.
Un atomo isolato ha uno spettro energetico discreto. Quando gli atomi si avvicinano tra loro, a partire da una certa distanza interatomica, i livelli energetici si dividono in zone. I livelli esterni vengono suddivisi per primi: vacanti 3 R, quindi riempito a metà il livello 3 S. Man mano che la distanza diminuisce R A R Si verifica 1 sovrapposizione 3 P- e 3 S-zone di energie consentite. A distanza r = r 0 (R 0 è la distanza interatomica di equilibrio nel cristallo), l'avvicinamento degli atomi si arresta. Valenza 3 S gli elettroni possono occupare qualsiasi stato all'interno di questa banda. Livelli 1 S e 2 S può dividersi solo quando R< r 0 e dentro legame chimico non partecipare. La connessione viene effettuata da un collettivo di elettroni di valenza, i cui stati energetici formano una banda comune risultante dalla sovrapposizione.
Nella zona delle energie consentite formate dai livelli di valenza, ce ne saranno 8 N stati (numero S-stati 2 N; numero R-stati 6 N). L'atomo ha un elettrone di valenza, quindi questa zona conterrà N elettroni che occupano stati secondo il principio di Pauli e il principio di minima energia. Di conseguenza, alcuni stati della zona sono liberi.
I cristalli in cui la zona formata dai livelli di elettroni di valenza è parzialmente riempita sono classificati come metalli Questa zona è chiamata zona di conduzione.
Semiconduttori e dielettrici
Consideriamo la struttura energetica dei semiconduttori e dei dielettrici usando l'esempio di un tipico semiconduttore: silicio cristallino (Z = 14), la cui formula elettronica è . Durante la formazione di un reticolo cristallino, a partire da una certa distanza interatomica R 1 >r 0 (R 0 – distanza interatomica di equilibrio nel cristallo). sp 3 -ibridazione degli stati elettronici del silicio, che non porta semplicemente alla sovrapposizione 3 S e 3 R zone, e alla loro fusione e formazione di un unico 3 sp 3 banda di valenza ibrida (Fig. 59), in cui il numero massimo possibile di elettroni è 8 N. Nel silicio cristallino, ogni atomo forma 4 legami tetraedrici, completando il suo guscio di valenza con un massimo di otto elettroni. Di conseguenza, nella banda di valenza tutti e 8 N gli stati sono occupati. Quindi, per semiconduttori e dielettrici banda formata dai livelli di elettroni di valenza- banda di valenza (VB) - completamente riempita. Prossimo posto vacante 4 S La banda -non si sovrappone alla banda di valenza a distanza interatomica R 0, ed è separato da esso da una zona di energie proibite (ZZ) . Gli elettroni situati nella banda di valenza non possono partecipare alla conduzione poiché tutti gli stati della banda sono occupati. Affinché nel cristallo appaia una corrente, è necessario trasferire gli elettroni dalla banda di valenza alla successiva banda libera di energie consentite. La prima zona libera consentita situata sopra si chiama banda di valenza zona di conduzione (CB). Viene chiamato lo spazio energetico tra la parte inferiore della banda di conduzione e la parte superiore della banda di valenza gap di banda Wg.
A seconda della larghezza del gap di banda, tutto corpi cristallini sono divisi in tre classi:
1. metalli - 0,1 eV;
2. semiconduttori -;
3. dielettrici - 4 eV.
Di conseguenza, i corpi hanno i seguenti valori di resistività:
1. metalli - ρ = 10 -8 10 -6 Ohm m;
2. semiconduttori - ρ = 10 -6 10 8 Ohm m;
3. dielettrici - ρ >10 8 Ohm m.
A temperatura T= 0 i semiconduttori sono dielettrici, ma con l'aumentare della temperatura la loro resistenza diminuisce drasticamente. Quando riscaldati, i dielettrici fondono prima che si verifichi la conduttività elettronica.
Uno dei problemi principali della teoria dello stato solido è determinare lo spettro energetico e gli stati stazionari degli elettroni in un cristallo. Un'idea qualitativa di questo spettro può essere ottenuta utilizzando metodi approssimati e semplificazioni. In primo luogo, si ritiene che il sottosistema dei nuclei sia praticamente a riposo (rispetto al rapido movimento degli elettroni) - approssimazione adiabatica. In secondo luogo, si presuppone che ciascun elettrone si muova in un campo creato da altri elettroni e indipendente dalla posizione istantanea di un dato elettrone, il che consente di considerare il movimento di ciascun elettrone indipendentemente da tutti gli altri e descritto dal sistema di Schrödinger a un elettrone. equazione.
Questa approssimazione si chiama singolo elettrone.
Approssimazione degli elettroni strettamente legati. In un atomo isolato, gli elettroni possono occupare solo livelli energetici discreti, separati da intervalli di energie proibite. In questo caso gli elettroni tendono ad occupare i livelli più bassi, ma a condizione che non vi siano più di due elettroni per ogni livello (principio di Pauli).
Durante la formazione di un cristallo, a causa dell'accostamento di N atomi identici, si creano tra loro forze di interazione: forze repulsive tra i nuclei e tra gli elettroni degli atomi vicini e forze di trazione tra tutti i nuclei e tutti gli elettroni.
L'approssimazione dell'elettrone fortemente legato si basa sull'idea che gli elettroni generalizzati mantengono una connessione sufficientemente forte con gli atomi e la loro energia potenziale può essere rappresentata nella forma seguente.
 , (4.20)
, (4.20)
Dove Ua– energia potenziale di un elettrone in un atomo isolato. Per un cristallo è una funzione periodica con periodo pari al parametro reticolare, poiché l'energia dell'elettrone si ripete quando passa da un atomo all'altro;  è un termine correttivo che tiene conto dell'influenza degli atomi vicini su questa energia.
è un termine correttivo che tiene conto dell'influenza degli atomi vicini su questa energia.
Se nella (4.20) trascuriamo il termine di correzione  ,quelli. considerare il cosiddetto approssimazione nulla, allora come funzione d'onda ed energia dell'elettrone nel cristallo dovremmo prendere la funzione d'onda
,quelli. considerare il cosiddetto approssimazione nulla, allora come funzione d'onda ed energia dell'elettrone nel cristallo dovremmo prendere la funzione d'onda  ed energia E a di un elettrone in un atomo isolato:
ed energia E a di un elettrone in un atomo isolato:  ,
, .
.
La differenza tra un cristallo e un singolo atomo in questo caso è la seguente. Mentre in un atomo isolato questo livello energetico E a è unico, in un cristallo costituito da N atomi si ripete N volte. In altre parole, ogni livello di un atomo isolato in un cristallo risulta esserlo N-moltiplicare degenere.
Consideriamo ora il termine di correzione  in energia potenziale (4.20). Quando gli atomi isolati si avvicinano e formano un reticolo, ogni atomo si ritrova in un campo sempre crescente di vicini con cui interagisce.
in energia potenziale (4.20). Quando gli atomi isolati si avvicinano e formano un reticolo, ogni atomo si ritrova in un campo sempre crescente di vicini con cui interagisce.
Nel campo di queste forze viene rimossa la degenerazione dei livelli atomici. Ecco perché ogni livello energetico, non degenere in un atomo isolato, si divide in N vicini sottolivelli situati l'uno dall'altro, formando una zona energetica. Questa zona è costituita da livelli energetici molto ravvicinati, la cui densità aumenta con la distanza dai bordi della zona secondo una legge parabolica, raggiungendo il massimo al centro della zona. Man mano che gli atomi si avvicinano, i livelli energetici più alti si dividono per primi, poi quelli più bassi man mano che gli atomi si avvicinano.
Il meccanismo di formazione delle zone energetiche è mostrato in Fig. 4.3.

Riso. 4.3. Schema di formazione delle bande energetiche in un cristallo
Se il livello di energia fosse in un atomo (2 l+1) –degenerazione multipla, allora sarà costituita la banda energetica corrispondente N(2 l+1) sottolivelli. Pertanto, il livello s fornisce la zona S, costituita da N sottolivelli e capace di ospitare 2 N elettroni: il livello p dà una banda p composta da 3 N sottolivelli e in grado di contenere 6N elettroni, ecc.
Poiché un cristallo con un volume di 1 m 3 contiene circa 10 28 atomi e la larghezza della banda energetica è di circa 1 eV, la distanza tra i livelli energetici nella banda è di circa 10–28 eV. Pertanto è sufficiente un impatto energetico trascurabile per provocare la transizione degli elettroni da un livello all'altro all'interno della banda; possiamo supporre che le bande di energia siano quasi continue.
L'influenza del campo reticolare sui diversi livelli dell'atomo non è la stessa. I livelli degli elettroni interni, che interagiscono fortemente con il nucleo, subiscono una scissione così debole che può essere trascurata: man mano che ci spostiamo verso sempre più elettroni esterni, l'energia della loro interazione con il nucleo diminuisce e l'influenza del campo esterno aumenta. Il cambiamento più forte sotto l'influenza del campo è sperimentato dai livelli degli elettroni di valenza esterni, che sono relativamente debolmente associati al nucleo, e zone energetiche, formato dai livelli energetici di questi elettroni, risulta essere il più ampio. Ciò è evidenziato dalla natura delle nubi elettroniche degli elettroni di valenza: si sovrappongono così tanto da creare una nuvola risultante di densità quasi uniforme. Ciò corrisponde allo stato della loro completa socializzazione nel reticolo. Tali elettroni socializzati vengono solitamente chiamati gratuito, e la loro totalità – elettronico gas.
Gli elettroni interni, fortemente legati al nucleo, subiscono solo lievi perturbazioni da parte degli atomi vicini, per cui il loro livello di energia nel cristallo rimane quasi altrettanto stretto che negli atomi isolati.
Pertanto, ogni livello energetico di un atomo isolato in un cristallo corrisponde a zona energetica consentita: livello 1 S – zona 1 S, livello 2 R– zona 2 R ecc. Le zone di energie consentite sono separate da aree di energie proibite – zone vietate E G. All’aumentare dell’energia degli elettroni in un atomo, l’ampiezza delle bande consentite aumenta, mentre l’ampiezza delle bande proibite diminuisce.
In molti casi, potrebbero esserci sovrapposizioni tra le aree consentite. Come i livelli energetici negli atomi isolati, le bande energetiche possono essere completamente piene di elettroni, parzialmente piene o vuote. Tutto dipende dalla struttura dei gusci elettronici degli atomi isolati e dalle distanze interatomiche nel cristallo. Viene chiamata la zona più alta, parzialmente o completamente piena di elettroni banda di valenza, e la zona vuota più vicina ad essa lo è zona di conduzione.
Approssimazione degli elettroni liberi. Consideriamo il caso del moto di un elettrone completamente libero lungo l'asse X, descritto come segue Equazione di Schrödenger:
 , (4.21)
, (4.21)
 , (4.22)
, (4.22)
poiché un elettrone libero ha energia cinetica.
La formula (4.22) rappresenta relazione di dispersione per gli elettroni liberi, che esprime la dipendenza E(p). Può essere convertito come segue. Secondo la formula di Louis de Broglie,
 , (4.23)
, (4.23)
dove λ è la lunghezza d'onda dell'elettrone e
 . (4.24)
. (4.24)
Il vettore k, in direzione coincidente con la direzione di propagazione dell'onda elettronica, e di grandezza pari a 2π/λ, è chiamato vettore d'onda dell'elettrone. Sostituendo p dalla (4.23) nella (4.22) otteniamo
 . (4.25)
. (4.25)
Dalle (4.22) e (4.24) è chiaro che per gli elettroni liberi la legge di dispersione è di natura quadratica e per il movimento unidimensionale degli elettroni è espresso da una parabola quadrata mostrata in Fig. 4.4.
La soluzione dell'equazione (4.21) è un'onda piana che viaggia:
 , (4.26)
, (4.26)
Dove UN– ampiezza dell'onda.

Fig.4.4. Legge di dispersione per un elettrone libero
Il modulo quadrato della funzione d'onda è proporzionale, come è noto, alla probabilità di rilevare un elettrone in una particolare regione dello spazio. Come si vede dalla (4.26), per un elettrone libero questa probabilità non dipende dalla coordinata dell'elettrone, poiché
Ciò significa che per un elettrone libero tutti i punti dello spazio sono equivalenti e la probabilità di trovarlo in qualcuno di essi è la stessa.
Approssimazione degli elettroni debolmente legati. Passiamo al caso del movimento degli elettroni in un campo periodico di un cristallo formato da ioni reticolari posizionati regolarmente (Fig. 4.5).

Fig. 4.5 Probabilità di rilevare un elettrone quando si muove in un campo di ioni posizionati correttamente
In questa approssimazione, l'energia potenziale di un elettrone è rappresentata come
; , (4.28)
, (4.28)
Dove U 0 (X) – energia potenziale di un elettrone nel campo degli ioni positivi, supponendo che questo campo sia compensato dal campo di tutti gli altri elettroni;
U 0 (X) - funzione periodica con periodo pari alla costante reticolare;
 - tiene conto della compensazione locale incompleta del campo ionico da parte degli elettroni. La probabilità di trovare un elettrone in una data posizione nel cristallo deve essere una funzione periodica della coordinata x, poiché le posizioni differiscono l'una dall'altra di una quantità che è un multiplo della costante reticolare UN(ad esempio, disposizioni A, A' E IN in Fig. 4.5) per un elettrone sono ugualmente probabili. Solo le posizioni all'interno dello stesso periodo saranno diverse UN(ad esempio, entro il periodo ACA). Questo significa questo ampiezza della funzione d'onda
- tiene conto della compensazione locale incompleta del campo ionico da parte degli elettroni. La probabilità di trovare un elettrone in una data posizione nel cristallo deve essere una funzione periodica della coordinata x, poiché le posizioni differiscono l'una dall'altra di una quantità che è un multiplo della costante reticolare UN(ad esempio, disposizioni A, A' E IN in Fig. 4.5) per un elettrone sono ugualmente probabili. Solo le posizioni all'interno dello stesso periodo saranno diverse UN(ad esempio, entro il periodo ACA). Questo significa questo ampiezza della funzione d'onda  l'elettrone che si muove in un campo periodico non rimane costante, come un elettrone libero, ma cambia periodicamente o, come si suol dire, modula con un periodo pari al periodo reticolare a. Indichiamo questa ampiezza con u(x). Quindi la funzione d'onda per un elettrone che si muove in un campo periodico cristallo nella direzione dell'asse x può essere rappresentato nella forma seguente:
l'elettrone che si muove in un campo periodico non rimane costante, come un elettrone libero, ma cambia periodicamente o, come si suol dire, modula con un periodo pari al periodo reticolare a. Indichiamo questa ampiezza con u(x). Quindi la funzione d'onda per un elettrone che si muove in un campo periodico cristallo nella direzione dell'asse x può essere rappresentato nella forma seguente:
 , (4.29)
, (4.29)
in questo caso u(x+na)=u(x), dove n è un numero intero qualsiasi. Viene richiamata la relazione (4.29). Funzione Bloch. La forma specifica di questa funzione è determinata dal tipo di energia potenziale U(x) inclusa nell'equazione di Schrödinger (4.9).
Di conseguenza, anche la relazione di dispersione degli elettroni che si muovono nel campo periodico del cristallo dovrebbe cambiare rispetto agli elettroni liberi. Innanzitutto, lo spettro energetico di tali elettroni acquisisce un carattere di banda. All'interno di ciascuna zona, l'energia degli elettroni risulta essere una funzione periodica del vettore d'onda k e per un cristallo unidimensionale (catena atomica) con il parametro UN può essere espresso dalla seguente relazione:
Dove E UN– energia del livello atomico da cui si è formata la zona; CON– spostamento di questo livello sotto l’influenza del campo degli atomi vicini; UN- cosiddetto integrale di scambio, tenendo conto della possibilità di transizione da atomo ad atomo che si è presentata per gli elettroni del cristallo a causa della sovrapposizione delle loro funzioni d'onda. Maggiore è la sovrapposizione tra le funzioni d'onda, cioè maggiore è la frequenza con cui gli atomi vicini possono scambiare i loro elettroni. Per gli stati s UN S <0 , per gli stati p UN P >0 , pertanto è consigliabile scrivere la relazione (4.30) separatamente per le zone s e p:
per le zone R
Dove  ;
; ;
; ,
, - valore assoluto degli integrali di scambio per questi stati.
- valore assoluto degli integrali di scambio per questi stati.
Nella fig. 4.6. sono mostrate le curve di dispersione E(k) per le zone s e p, costruzione utilizzando le equazioni (4.31) e (4.32).
Per gli stati s E s at k=0
assume il valore minimo  . All'aumentare k, diminuisce coska E
. All'aumentare k, diminuisce coska E  cresce, raggiungendo il suo valore massimo
cresce, raggiungendo il suo valore massimo  A
A  .
.

Fig.4.6. Dipendenza E(k) nella rappresentazione delle zone date
Cambia allo stesso modo E S (k) quando si cambia k da 0 A - π/a. Larghezza della zona s consentita che si estende da E S min A E S Massimo, è uguale
Per gli stati p  si trova a
si trova a 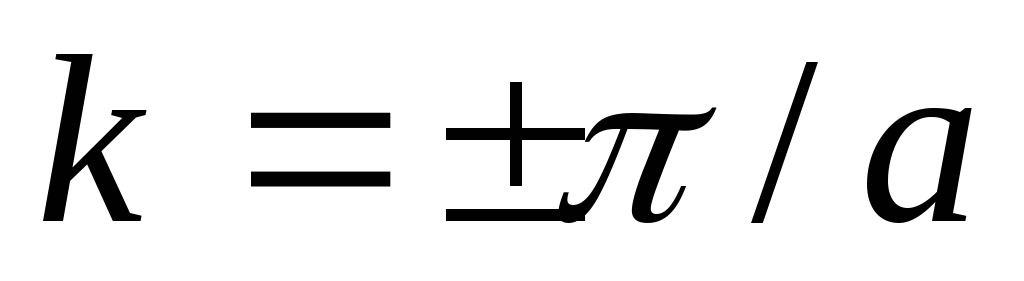 , UN
, UN  a k=0. Larghezza della zona P
a k=0. Larghezza della zona P
è ancora determinato dal valore dell'integrale di scambio A r. Di norma, quanto più alto è il livello atomico, tanto più si sovrappongono le funzioni d'onda degli elettroni di questo livello nel cristallo, tanto maggiore è l'integrale di scambio e più ampia la banda energetica formata da questo livello. Pertanto, a partire da livelli atomici elevati, si formano ampie bande di energia, separate da stretti gap di banda (vedi Fig. 4.3).
Intervalli di vettori d'onda k, all'interno del quale l'energia E(k)
l'elettrone, come funzione periodica k, sperimenta un ciclo completo del suo cambiamento, chiamato Zone di Brillouin. Per un cristallo unidimensionale (catena atomica), la prima zona di Brillouin si estende da  A
A  e ha una lunghezza
e ha una lunghezza  (Fig. 4.6), due segmenti da
(Fig. 4.6), due segmenti da  A
A  e da
e da  A
A  rappresentano la seconda zona di Brillouin, ecc. Con valori
rappresentano la seconda zona di Brillouin, ecc. Con valori  , Dove
, Dove  l'energia subisce una rottura, portando alla formazione di band gap di ampiezza E g .
l'energia subisce una rottura, portando alla formazione di band gap di ampiezza E g .
Tutti i possibili valori energetici in ciascuna zona energetica possono essere ottenuti modificando k all'interno della prima zona di Brillouin, quindi la dipendenza E(k) spesso costruito solo per la prima zona. Tutti gli altri valori E possono essere portati in questa zona. Questo modo di rappresentare E(k) chiamato diagramma delle zone indicate(Fig. 4.6). È possibile un altro metodo, chiamato schema delle zone estese (Fig. 4.7).
Qui si trovano le diverse zone energetiche k-spazi in diverse zone di Brillouin.

Fig.4.7. Rappresentazione delle bande energetiche in un diagramma a bande estese
Nella fig. 4.7 mostra anche la dipendenza parabolica E(k)
per un elettrone libero. Vicino agli estremi della curva di dispersione, cioè punti vicini k=0
E  (centro e confine della prima zona di Brillouin),
(centro e confine della prima zona di Brillouin),  possono essere disposti in fila secondo ka
(k contare da 0 se l'estremo è al centro della zona di Brillouin, e da
possono essere disposti in fila secondo ka
(k contare da 0 se l'estremo è al centro della zona di Brillouin, e da  , se l'estremo è al confine della zona di Brillouin) e limitarci ai primi due termini dello sviluppo:
, se l'estremo è al confine della zona di Brillouin) e limitarci ai primi due termini dello sviluppo:
Sostituendo ciò nelle (4.31) e (4.32) otteniamo:
Il minimo della curva di dispersione si chiama E(k). fondo della zona energetica, massimo – superiore O soffitto zone. Pertanto le relazioni ottenute possono essere riscritte nella seguente forma più generale:
Per il fondo della zona;
Per la zona del soffitto.
Pertanto, all'estremità inferiore e superiore della banda energetica, l'energia degli elettroni è proporzionale al quadrato del vettore d'onda, calcolato nel modo sopra descritto, e all'integrale di scambio, che determina l'ampiezza della banda. Nella Fig. 4.6, le parabole corrispondenti alle equazioni 4.35 e 4.36 sono mostrate con una linea tratteggiata.
Consideriamo la natura fisica delle discontinuità nello spettro energetico dell'elettrone ai confini delle zone di Brillouin. Esprimiamoci k attraverso la lunghezza d'onda dell'elettrone λ e scrivere la condizione per la discontinuità della funzione E(k):
O  . (4.37)
. (4.37)
Questa è la ben nota condizione di Wulff-Bragg per un'onda di elettroni incidente su un reticolo perpendicolare ai piani atomici. Di conseguenza, le discontinuità nello spettro energetico di un elettrone in un cristallo si verificano quando è soddisfatta la condizione di riflessione di Bragg (4.37). Gli elettroni con questa lunghezza d'onda subiscono la riflessione e non possono propagarsi nel cristallo.
Per i cristalli reali, la dipendenza E(k) è, di regola, molto più complessa di quella descritta dalla formula (4.30).
Nella fig. 4.8. A titolo di esempio, sono mostrate le curve di dispersione che limitano la banda di conduzione (curva 1) e la banda di valenza (curva 2) del silicio.

Riso. 4.8. Curve di dispersione e diagramma a bande del silicio
Per facilitare la presentazione, continueremo la discussione utilizzando l'esempio particolare di una particella con massa in presenza di un potenziale scalare. Supponiamo anche che la Funzione dipenda dal vettore che fissa la posizione della particella e l'equazione di Schrödinger , indipendente da
tempo, sarà scritto nel modulo
Nel linguaggio della teoria delle equazioni alle derivate parziali, un'equazione come la (36) è chiamata equazione agli autovalori. La soluzione di questa equazione è un'autofunzione corrispondente all'autovalore E dell'operatore H.
In realtà il problema degli autovalori si definisce solo se si stabiliscono le condizioni di “regolarità” e le condizioni al contorno che la funzione deve soddisfare. Le condizioni imposte alla funzione devono, ovviamente, essere coerenti con l'interpretazione generale della funzione d'onda. Torneremo su questo argomento nel cap. IV. Richiediamo qui che la funzione e le sue derivate parziali del primo ordine siano continue e funzioni limitate in tutto lo spazio.
In questo caso è possibile dimostrare la validità dei seguenti risultati, che accetteremo come dati, ma potremo verificarli utilizzando numerosi esempi.
a) Se allora l'equazione (36) ha soluzioni solo per certi determinati valori di E, formando uno spettro discreto. L'autofunzione per qualsiasi autovalore (o ciascuna funzione, se ce ne sono più) svanisce all'infinito. Più precisamente, converge l’integrale esteso all’intero spazio delle configurazioni. Secondo l'interpretazione statistica, ciò significa che la probabilità di trovare una particella all'infinito è zero; la particella rimane localizzata in una regione finita dello spazio; Si dice che la particella sia in uno stato legato.
b) Se allora l'equazione (36) può avere soluzioni per qualsiasi valore positivo di E. Dicono che le energie positive formano uno spettro continuo. Le autofunzioni corrispondenti non si annullano all'infinito; il loro comportamento asintotico è simile a quello di un'onda piana. Più precisamente il modulo tende ad una costante finita oppure oscilla tra valori, di cui almeno uno è diverso da zero. La particella non rimane localizzata nella regione finita. Funzioni d'onda di questo tipo vengono utilizzate per descrivere problemi di collisione; dicono che abbiamo a che fare con una particella in uno stato non legato, o in stato stazionario dispersione.
Otteniamo così il primo risultato fondamentale: la quantizzazione dei livelli energetici degli stati legati, ovvero uno dei fatti sperimentali più impressionanti,
causato lo schianto teoria classica. La determinazione dei livelli energetici quantizzati viene qui presentata come un problema di ricerca degli autovalori. Risolvere questo problema con il massimo grado di precisione possibile è uno dei problemi centrali della meccanica ondulatoria. Per alcuni soprattutto forme semplici Il problema hamiltoniano può essere risolto rigorosamente. Questo è proprio il caso dell’atomo di idrogeno (lo considereremo in dettaglio nel Capitolo XI), quando i livelli energetici risultano essere gli autovalori dell’operatore. Lo spettro risultante coincide con quello previsto dalla vecchia teoria quantistica ; Abbiamo già avuto occasione di sottolineare la sorprendente coincidenza di questo spettro con i dati sperimentali. Di più situazioni difficili dovrebbero essere utilizzati vari metodi approssimativi. Ma in tutti i casi in cui è stato possibile calcolare lo spettro energetico con un sufficiente grado di precisione, l'accordo con l'esperimento si è rivelato il migliore che ci si poteva aspettare da una teoria non relativistica.
La propria funzione stessa può essere soggetta in una certa misura verifica sperimentale. Infatti, le autofunzioni di uno spettro discreto vengono utilizzate nel calcolo di varie quantità osservabili, ad esempio le probabilità delle transizioni quantistiche. Per quanto riguarda le autofunzioni dello spettro continuo, la loro forma asintotica è direttamente correlata alle sezioni d'urto efficaci che caratterizzano i fenomeni di scattering, che verranno chiariti in dettaglio in seguito. Nel campo della fisica atomica non relativistica non è stato ancora scoperto un solo caso di discrepanza tra le previsioni della meccanica ondulatoria e i dati sperimentali.
proporzionale a T: n ~T. Di conseguenza, il coefficiente di conduttività termica dovrebbe essere inversamente proporzionale alla temperatura, il che è in accordo qualitativo con l'esperimento. A temperature inferiori alla temperatura di Debye, l è praticamente indipendente da T, e la conduttività termica è interamente determinata dalla dipendenza della capacità termica del cristallo da T, CV ~ T 3. Pertanto a basse temperature λ ~T 3. La dipendenza caratteristica della conduttività termica dalla temperatura è presentata nella Figura 9.
Nei metalli, oltre alla conduttività termica reticolare, è necessario tenere conto anche della conduttività termica dovuta al trasferimento di calore da parte degli elettroni liberi. Ciò spiega l'elevata conduttività termica dei metalli rispetto ai non metalli.
3. Struttura elettronica dei cristalli.
3.1.Movimento degli elettroni in un campo periodico. Struttura a bande dello spettro energetico degli elettroni in un cristallo. Funzioni di Bloch. Curve di dispersione. Massa effettiva.
In un solido, le distanze tra gli atomi sono paragonabili alle loro dimensioni. Pertanto, i gusci elettronici degli atomi vicini si sovrappongono parzialmente l'uno all'altro e almeno gli elettroni di valenza di ciascun atomo si trovano in un campo abbastanza forte di atomi vicini. Una descrizione accurata del movimento di tutti gli elettroni, tenendo conto dell'interazione coulombiana degli elettroni tra loro e con i nuclei atomici, è un compito estremamente difficile anche per un singolo atomo. Pertanto, viene solitamente utilizzato il metodo del campo autoconsistente, in cui il problema si riduce alla descrizione del movimento di ogni singolo elettrone nel campo potenziale effettivo creato dai nuclei atomici e del campo medio degli elettroni rimanenti.
Consideriamo innanzitutto la struttura dei livelli energetici di un cristallo, basata sull'approssimazione di legame forte, in cui si assume che l'energia di legame di un elettrone con il suo atomo superi significativamente energia cinetica il suo movimento da atomo ad atomo. A grandi distanze tra gli atomi, ciascuno di essi ha un sistema di stretti livelli energetici corrispondenti stati correlati elettrone con ione. Man mano che gli atomi si avvicinano, la larghezza e l'altezza delle barriere potenziali tra loro diminuiscono e, grazie all'effetto tunnel, gli elettroni sono in grado di spostarsi da
da un atomo all'altro, che è accompagnato da un'espansione dei livelli energetici e dalla loro trasformazione in zone energetiche.(Fig. 10). Ciò è particolarmente vero per gli elettroni di valenza debolmente legati, che sono in grado di spostarsi facilmente attraverso il cristallo da un atomo all'altro e in una certa misura diventano simili agli elettroni liberi. Gli elettroni dei livelli energetici più profondi sono ciascuno molto più fortemente legati al proprio atomo. Formano bande energetiche strette con ampie gamme di energie proibite. Nella fig. La Figura 10 mostra convenzionalmente le curve potenziali e i livelli di energia per un cristallo di Na. La natura generale dello spettro energetico degli elettroni in funzione della distanza internucleare, d, è presentata nella Figura 11. In alcuni casi, i livelli superiori si allargano così tanto che le bande energetiche adiacenti si sovrappongono. Nella fig. 11 ciò si verifica quando d = d1.
Sulla base della relazione di incertezza di Heisenberg-Bohr, l'ampiezza della banda di energia, ∆ε, è correlata al tempo di residenza τ di un elettrone in un determinato sito del reticolo dalla relazione: ∆ε τ > h. A causa dell'effetto tunnel, un elettrone può fuoriuscire attraverso una barriera di potenziale. Secondo la stima, alla distanza interatomica d ~ 1Aτ ~ 10 -15 s, e quindi ∆ε ~ h/τ ~ 10 -19 J ~ 1 eV, cioè Il gap di banda è dell'ordine di uno o più eV. Se un cristallo è costituito da N atomi, ciascuna banda energetica è composta da N sottolivelli. Un cristallo di 1 cm3 contiene N~ 1022 atomi. Di conseguenza, con una larghezza di banda di ~ 1 eV, la distanza tra i sottolivelli è di ~ 10 -22 eV, il che è significativo meno energia movimento termico in condizioni normali. Questa distanza è così insignificante che nella maggior parte dei casi le zone possono essere considerate praticamente continue.
In un cristallo ideale, i nuclei atomici si trovano ai nodi del reticolo cristallino, formando una struttura strettamente periodica. Di conseguenza anche l'energia potenziale dell'elettrone, V(r), dipende periodicamente dalle coordinate spaziali, cioè ha simmetria traslazionale:
reticoli, a i (i = 1,2,3,...) – vettori delle principali traslazioni.
Le funzioni d'onda e i livelli di energia in un campo periodico (1) vengono determinati risolvendo l'equazione di Schrödinger
che rappresenta il prodotto dell'equazione di un'onda piana, ei kr, per un fattore periodico, u k (r) = u k (r + a n), con il periodo reticolare. Le funzioni (3) sono chiamate funzioni di Bloch.
Per V(r) = 0 l'equazione (2) ha soluzione sotto forma di onda piana:
dove m è la massa della particella. Viene rappresentata la dipendenza dell'energia E dal numero d'onda curva di dispersione. Secondo (5), nel caso di un elettrone libero, questa è una parabola. Per analogia con il moto libero, il vettorek nell'equazione (3) è chiamato vettore d'onda, аp = h k – quasi-momento.
Nell'approssimazione dell'accoppiamento debole si considera il movimento degli elettroni quasi liberi, che sono influenzati dal campo perturbatore del potenziale periodico dei nuclei ionici. A differenza del moto libero, in un campo periodico V(r) l’equazione (2) non ha soluzioni per tutti i valori di E. Aree di energie consentite si alternano ad aree di energie proibite. Nel modello di accoppiamento debole, ciò è spiegato dalla riflessione di Bragg delle onde degli elettroni nel cristallo.
Consideriamo questo problema in modo più dettagliato. La condizione per la massima riflessione delle onde elettroniche in un cristallo (condizione di Wulff-Bragg) è determinata dalla formula (17) parte I. Considerando che G = n g, otteniamo:
Consideriamo un sistema di intervalli finiti che non contengono valori di k che soddisfano la relazione (7):
( - n g /2 La regione di variazione di k nello spazio k tridimensionale, data dalla formula (8) per tutte le direzioni possibili, determina i confini dell'ennesima zona di Brillouin. All'interno di ciascuna zona di Brillouin (n= 1,2,3,...) l'energia degli elettroni è una funzione continua di k, e ai confini delle zone subisce una discontinuità. Infatti, se la condizione (7) è soddisfatta per l’ampiezza dell’incidente, ψ k (r) = uk (r) ei kr e riflesso ψ -k (r) = u - k (r) e -i kr le onde saranno le stesse, u k (r) = u -k (r). Queste onde danno due soluzioni all'equazione di Schrödinger: Questa funzione descrive l'accumulo di carica negativa sugli ioni positivi, dove l'energia potenziale è più bassa. Analogamente, dalla formula (9b) si ottiene: ρ 2 (r) = |ψ 2 (r)|2 =4 u g/2 2 (r)sen 2 (gr/2) Questa funzione descrive una distribuzione degli elettroni in cui essi si trovano prevalentemente in aree corrispondenti alla metà delle distanze tra gli ioni. In questo caso l’energia potenziale sarà maggiore. La funzione ψ 2 corrisponderà all'energia E2 > E1. spazi tra le bande di larghezza Ad es. L'energia E`1 determina il confine superiore della prima zona e l'energia E2 determina il confine inferiore della seconda zona. Ciò significa che quando le onde degli elettroni si propagano nei cristalli si creano intervalli di energia per i quali non esistono soluzioni dell'equazione di Schrödinger che abbiano natura ondulatoria. Poiché la natura della dipendenza dell'energia dal vettore d'onda influenza in modo significativo la dinamica degli elettroni in un cristallo, è interessante considerare, ad esempio, il caso più semplice di una catena lineare di atomi situati ad una distanza a l'uno dall'altro lungo l'asse x. In questo caso g = 2π /a. La Figura 12 mostra le curve di dispersione per le prime tre zone Brillouin unidimensionali: (- π/a< k <π
/a), (-2π
/a < k < -π
/a; π/
a < k < 2π
/a), (-3π/
a < k < -2π
/a; 2π
/a < k < 3π
/a). К запрещенным зонам относятся области энергии Е`1
< E < E2
, E`2
< E< E3
и т.д. Nella fig. 12 presentati circuito di zona estesa, in cui diverse zone energetiche si trovano nello spazio VC in diverse zone di Brillouin. Tuttavia è sempre possibile, e spesso conveniente, scegliere un vettore d'onda in modo che la sua estremità si trovi all'interno della prima zona di Brillouin. Scriviamo la funzione di Bloch come: si trovano nella prima zona di Brillouin. Sostituendo k nella formula (11), otteniamo: ha la forma di una funzione di Bloch con il moltiplicatore di Bloch (13). L'indice n ora indica il numero della zona energetica a cui appartiene la funzione data. Viene chiamata la procedura per portare un vettore d'onda arbitrario nella prima zona di Brillouin diagrammi delle zone indicate. In questo schema vectork assume i valori -g/2< k < g/2
, но одному и тому же значениюк
будут отвечать различные значения энергии, каждое из которых будет соответствовать одной из зон. На рисунке 13 представлена схема приведенных зон для одномерной решетки, соответствующая расширенной зонной схеме на рисунке 12. Pertanto, l'esistenza di gap energetici è dovuta alla riflessione di Bragg delle onde elettroniche di de Broglie dai piani cristallini. I punti di rottura sono determinati dalle condizioni di massima riflessione delle onde. Secondo le leggi della meccanica quantistica, il moto traslatorio di un elettrone è considerato come il moto di un pacchetto d'onde con vettori d'onda vicini al vettore k. La velocità di gruppo del pacchetto d'onde, v , è data da